Tämä on ristikko! Alkuperäinen otettu eka_tyryshkina a Luonnonvirheet: ihmiset, joilla on harvinaisia \u200b\u200bsairauksia.
Sairasin täällä toisena päivänä, kuten aina, koska meidän on vain mentävä käymään, olen sairas! Huopa sairaudeni vastaa tapahtuman suunnitteluun, huopa vielä jotain, mutta on hyvä, että se ei reagoi työhön. Yleensä sairauteni ei ole yksinkertainen)
Ja nyt olin sairas kotona myöhään, olin jo suunnitellut kaikki asiat, lukenut uudelleen ja käynyt läpi kaikki mielenkiintoiset sivustot, en yhtäkkiä odottanut itseni saavan selville planeetan harvinaisimmista sairauksista ja tiedät niin paljon mielenkiintoista ja järkyttävää !!!
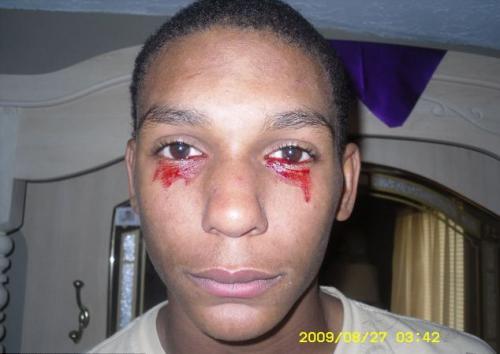
haemolacria
(”Verisiä kyyneleitä”) havaitaan yhdellä henkilöllä miljoonaa kohden. Veri kyynelnesteen sijasta alkaa virtata silmistä yhtäkkiä ja se voi kestää noin tunnin. Päivän aikana potilas irtoaa verisillä kyynelillä 3 - 20 kertaa.
Tämän taudin tarkkaa syytä ei ole täysin selvitetty, joten sitä ei voida parantaa. Lääketieteen asiantuntijat ovat toistaiseksi edenneet ajatukseen, että hemolakria on yksi veren tai kasvaimen sairauksista.
Kuvassa - 15-vuotias Calvino Inman (Tennessee, Yhdysvallat)

Vampire-oireyhtymä
Diagnoosilla "Vampire-oireyhtymä"
(ektodermaalinen dysplasia) maailmassa on vain 7 tuhatta ihmistä.
Kuolemaan johtavan vaalean ihon ja terävien hampaiden lisäksi (jos osaa hampaista ei ole), potilailla on harvinaisia \u200b\u200bja ohuita hiuksia, hikointikyky heikkenee, joten heidän ruumiinsa altistuu ylikuumenemiselle. Oireet ilmenevät lapsuudessa, mutta tauti voidaan havaita jo raskauden vaiheessa geenitesteillä.
Pojat pakotetaan käyttämään aurinkolaseja ja käyttämään aurinkovoidetta, kun he menevät ulos, koska he eivät voi olla suoran auringonvalon alla.Samalla fyysinen kehitys ja fyysinen aktiivisuus pysyvät normaalina.Itse tauti on parantumaton, vain oireet voidaan korjata. Erityisesti voit palauttaa hampaiden normaalin muodon.
Simonin tauti diagnosoitiin lapsenkengissä. Kun Mandy oli raskaana toisen kerran, häntä varoitettiin, että toisella lapsella voi olla sama sairaus.Simon kuitenkin kasvoi ja kehittyi hyvin, joten vanhemmat ottivat tämän riskin.
Pojat sanovat: "Jotkut lapset nauravat ulkonäköämme, mutta ystävämme mielestä se on siistiä".
Kuvassa - Simon (13-vuotias) ja George (11-vuotias) Cullen (Suffolk, Iso-Britannia).

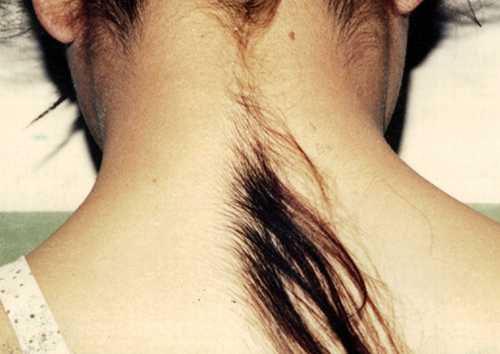
hypertrikoosi
("Ihmissusi-oireyhtymä") on sairaus, joka ilmenee liiallisessa hiuskasvussa, joka ei ole ominaista tälle ihoalueelle, ei vastaa sukupuolta ja ikää. Vain vähän yli neljäkymmentä tällaista potilasta on rekisteröity ympäri maailmaa, joten heille sopivin tapa ansaita rahaa on näyttää rumuus ... He jättävät hakemuksia Guinnessin ennätyskirjaan - tullakseen kuuluisiksi ja ansaitakseen rahaa ... Kiinalainen Yu Zhenhuang teki sen sataprosenttisesti - superkarvaisuutensa ansiosta hän perusti maansa suosituimman rock-yhtyeen ja tuli miljoonasta.
Ei ole tiedossa, miksi tämä mutaatio tapahtuu. Ja kukaan ei ole vielä kehittänyt hypertrichoosin hoitoa. Kosmetologit voivat poistaa hiuksia vain riittävän kauan ...
Kuvassa - 6-vuotias Nat Sasufan (Thaimaa), 2007

Kuvassa on 33-vuotias Yu Zhenhuang (Kiina), maailman karvaisin mies

Norsu tauti
(Proteus-oireyhtymä, elephantiasis, elephantiasis, elephantiasis) - minkä tahansa kehon osan suurentuminen ihon ja ihonalaisen kudoksen kivullisen kasvun takia. Maailmassa on noin 120 ihmistä, joilla on tämä parantumaton sairaus ...
Ja tunnetuin potilas oli "norsu mies" - Joseph Merrick. Tietoja kuuluisasta britistä vuonna 1980, ohjaaja David Lynch jopa teki elokuvan, joka oli ehdolla Oscariksi kahdeksassa kategoriassa ... Elokuva oli ihmisarvosta ... Merrickiä pelannut John Hurt luotiin Lontoon kuninkaallisessa sairaalassa pidetyn esityksen perusteella. Joseph Merrickin alkoholipitoisuus. Hänen tyynynsä käytti näyttelijää päivittäin 12 tuntia päivässä ...
Kuvassa - 35-vuotias Mehndi Sellars (Iso-Britannia)

Nopeutettu geenin epänormaalisuus kehon ikääntyminen ,
- progeria - jaettu lastentarhaan (Getchinsonin oireyhtymä) ja aikuisiin (Wernerin oireyhtymä). Ensimmäistä kertaa he alkoivat puhua ennenaikaisen ikääntymisen oireyhtymästä 100 vuotta sitten. Ja ei ole yllättävää, että tällaisia \u200b\u200btapauksia esiintyy kerran 4–8 miljoonasta vastasyntyneestä. Progeria (kreikkalaisesta aikaisemmasta, gerontos-vanha mies) on erittäin harvinainen geneettinen sairaus, joka nopeuttaa ikääntymisprosessia noin 8-10 kertaa. Yksinkertaisesti sanottuna, lapsen ikä on 10–15 vuotta yhdessä vuodessa. Kahdeksanvuotias näyttää olevan 80-vuotias - kuivalla ryppyisellä iholla, kalju pää ... Nämä lapset kuolevat yleensä 13 - 14-vuotiaina useiden sydänkohtausten ja aivohalvausten jälkeen etenevän ateroskleroosin, kaihien, glaukooman, täydellisen hampaiden menetyksen jne. Taustalla. Ja vain harvat elävät jopa 20 vuotta tai kauemmin.
Nyt maailmassa tunnetaan vain 42 progeriatapausta ... Näistä 14 ihmistä asuu Yhdysvalloissa, 5 Venäjällä ja loput Euroopassa ...
Tällä hetkellä on useita organisaatioita, jotka tarjoavat apua pienille vanhuksille ja heidän perheilleen. Internetissä on tätä erityistä ongelmaa varten omistettuja verkkosivustoja, joista osa on avoin lääkäreille tai sosiaalityöntekijöille, toiset potilaiden perheille.
Kuvassa - 24-vuotias Leon Bot



38-vuotias puu mies
Dede Cosvarasta, joka asuu Java-saarella, Indonesiassa, on tullut maailmankuulu ihmisen papilloomaviruksesta, joka yleensä johtaa pienten syylien esiintymiseen, mutta Indonesian tapauksessa muotoutui tuntemattomasti raajoihinsa.
Deden ongelmana oli, että hänellä oli harvinainen geneettinen poikkeavuus, joka esti immuunijärjestelmäänsä estämästä näiden syylien kasvua. Siksi virus pystyi "ottamaan haltuunsa ihosolujensa solumekanismin", käskeen heitä tuottamaan suuren määrän kiimaista ainetta, josta he koostuivat. Dede löysi myös alhaisen valkosolujen määrän.


Perhostauti
Hyperplastisessa muodossa oleva bullous epidermolyysi on geneettinen sairaus, joka ilmenee ensimmäisinä elämänpäivinä. Itse asiassa vastasyntyneen iho on niin herkkä, että mikä tahansa kosketus johtaa haavojen ja rakkuloiden muodostumiseen. Vaikuttavat eniten ulkonevat alueet: kyynärpäät, polvet, jalat, kädet. Haavauma, jolla iho irtoaa kerroksittain, ei parane pitkään, neste vapautuu siitä. Suuren vadelmaarven muodostumisen jälkeen.
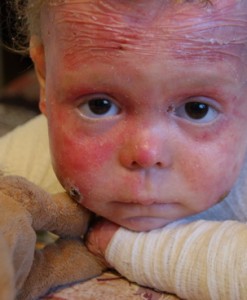
Tätä tautia ei voida parantaa, vain oireiden lievitys on mahdollista. Ei niin kauan sitten, koko Lisa Kunigelin tarina ukkosi kaikkialla Venäjällä, joka on asunut bulloosisessa epidermolyysissä melkein kymmenen vuotta. Hän tarvitsee useita kertoja päivässä sidoksia ja hoitoa antimikrobisilla voiteilla ja geeleillä. Lisäksi Lisaan on kaikkien 9 vuoden ajan liittynyt kipua.
Merenneito-oireyhtymä
Yksi kehityksen harvinaisimmista poikkeavuuksista on sirenomelia, jota kutsutaan nimellä "merenneito-oireyhtymä". Tämän vian myötä vastasyntyneet syntyvät silmukoiduilla jaloilla, jotka ovat samanlaisia \u200b\u200bkuin kalahäntä. Heillä on vain yksi munuainen toiminta, ei sukuelimiä. Koska laaja tappio sisäelimet sellaiset vauvat kuolevat yleensä pian. Tauti esiintyy yhdellä 100 000 vastasyntyneestä. Kaikkien havaintovuosien aikana vain kolme lasta pystyi selviytymään. Yksi heistä oli Shailo Pepin.
Shiloh syntyi vuonna 1999 ja hänestä tuli tunnetuin lapsi, jolla oli ”merenneito-oireyhtymä”. 10 vuoden aikana, jonka hän pystyi elämään, hän loi tuhansia ystäviä ympäri maailmaa, jotka tukivat tyttöä ja hänen äitinsä. Shiloh yritti elää täyden elämän - kuten kaikki tavalliset lapset, hän kävi koulussa, osallistui tanssitunneille, kävi huvipuistoissa. Tyttö tuli kuuluisaksi osallistuttuaan Oprah Winfrey -esitykseen. Learning Chanel teki useita elokuvia hänestä, sadat Internet-sivustot ovat omistettu hänelle.

Tarina Shilohin kanssa on uskomaton tarina ihmeestä. Lapsi, joka taisteli koko lapsuutensa selviytyäkseen. Pieni tyttö, joka tiesi kuinka nauttia joka päivä, parantumattomasta taudista huolimatta.
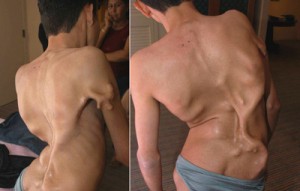
Münchenin tauti
Fibrodysplasia on erittäin harvinainen sairaus. Viralliset tilastot ovat seuraavat: 1 potilas / 2 000 000 ihmistä. Münheimerin tauti esiintyy geenimutaation seurauksena ja syntymän yhteydessä ilmenee ulkoisina vaurioina. Vauvan isot varpaat ja selkäranka ovat taipuneet. Patologia johtaa vammaisuuteen, varhaiseen kuolleisuuteen. Missä tulehduksen vastaisten prosessien pitäisi tapahtua, luun kasvu alkaa muodostua, minkä vuoksi tautia kutsutaan usein "toisen luurankon sairaudeksi".
Mikä tahansa, jopa vähäinen mustelma, voi johtaa lasitusten kehittymiseen kyseisellä alueella, mutta tällä hetkellä ei ole virallista hoitoa tappavalle taudille. Tutkijat ovat kehittäneet lääkkeen, joka voi teoreettisesti torjua tautia. Tarvittavia kliinisiä tutkimuksia ei kuitenkaan ole vielä tehty. Valitettavasti on erittäin vaikea toteuttaa niitä - kaikkialla maailmassa ei ole enempää kuin 600 Münheimerin tautia sairastavaa.
ilmiö "Lines Blashko" jolle on ominaista outojen bändien läsnäolo kehossa. Blashko-linjat ovat DNA: han upotettua näkymätöntä kuviota. Ja tämän mallin ulkonäöstä tulee taudin ilmentymä.
Tyypillisesti kuvio takana on V-muotoinen, rinnassa, vatsassa ja sivuilla - S-muotoinen.
Taudin syy voi olla mosaiikki. Joka tapauksessa Blashko-linjojen ulkonäkö ei ole millään tavoin yhteydessä ihmisen hermostoon, lihaksiin ja imusysteemeihin.
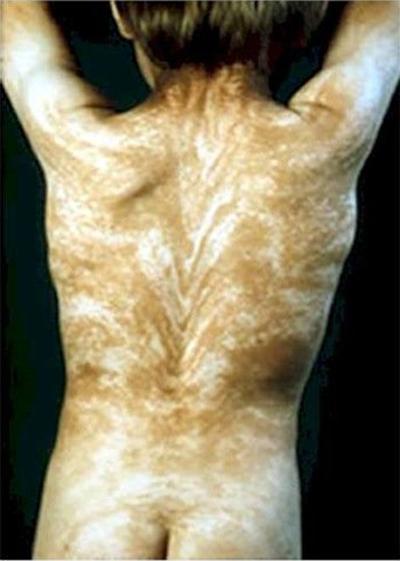
Toinen epänormaali sairaus - acanthosis nigricanstai "sinisen ihon oireyhtymä". Ihmisillä, joilla on tämä diagnoosi, voi olla sininen, indigo-, luumu- tai melkein violetti iho. Viime vuosisadan 60-luvulla Kentuckyssa asui koko “sinisten” ihmisten perhe. Ne tunnettiin sinisinä fugaateina. Tämä ominaisuus on siirretty sukupolvelta toiselle.


Noin 6% maailman asukkaista kärsii harvinaisista sairauksista, ja tämä määrä kasvaa edelleen. Kaikilla ainutlaatuisilla sairauksilla on erilainen luonne, mutta suurin osa ilmiömäisistä vaivoista liittyy geneettisiin poikkeavuuksiin ja infektioihin.
Geneettiset sairaudet ovat ainutlaatuisia siinä mielessä, että ne eivät ole riippuvaisia \u200b\u200bihmisen elämäntavasta, et voi vakuuttaa heitä vastaan \u200b\u200byksinkertaisesti lopettamalla syö rasvaisia \u200b\u200bruokia tai aloittamalla liikunnan aamulla. Ne syntyvät mutaatioiden seurauksena ja voivat siirtyä sukupolvelta toiselle.
Harvinainen perinnöllinen sairaus, jossa ihminen kuolee kyvyttömyydestä nukahtaa. Tähän asti sitä on juhlittu vain 40 perheessä ympäri maailmaa. Tappava unettomuus tapahtuu yleensä 30–60 vuoden aikana (useimmiten 50 vuoden jälkeen) ja kestää 7–36 kuukautta. Taudin edetessä potilas kärsii vakavista unihäiriöistä, eikä unilääkkeet auta häntä. Ensimmäisessä vaiheessa unettomuuteen liittyy paniikkikohtauksia ja fobioita, toisessa vaiheessa niihin lisätään hallusinaatioita ja lisääntynyttä hikoilua. Sairauden kolmannessa vaiheessa ihminen menettää kokonaan kykynsä nukkumaan ja alkaa näyttää paljon vanhemmalta kuin ikänsä. Sitten dementia kehittyy ja potilas kuolee - yleensä uupumasta tai keuhkokuumeesta.

Narkolepsia-katapleksioireyhtymä, jolle on ominaista äkilliset unen hyökkäykset ja kehon lihasrelaksaatio, on myös geneettinen luonne ja esiintyy unen nopean vaiheen häiriöiden takia. Sitä esiintyy paljon useammin kuin kohtalokasta unettomuutta: 40 sadasta tuhannesta ihmistä, samoin kuin miehillä ja naisilla. Narkolepsiasta kärsivä henkilö voi yhtäkkiä nukahtaa useita minuutteja keskellä päivää. ”Unihäiriöt” muistuttavat REM-vaihetta ja niitä voi tapahtua hyvin usein: jopa 100 kertaa päivässä, ennen niitä olevaa päänsärkyä tai ilman. Niitä provosoidaan usein toimimattomuudesta, mutta niitä voi tapahtua täysin sopimattomassa ajassa: yhdynnän aikana, urheilemassa, ajaessasi. Henkilö herää levätä.
![]()
Juner Tanin oireyhtymälle (SUT) on tunnusomaista ensisijaisesti se, että siitä kärsivät ihmiset kävelevät neljäsosassa. Turkkilainen biologi Juner Tan löysi sen tutkittuaan viittä Ulas-perheen jäsentä Turkin maaseudulla. Useimmiten SUT-potilaat käyttävät primitiivistä puhetta ja heillä on synnynnäinen aivojen vajaatoiminta. Vuonna 2006 kuvattiin Ulasin perheestä dokumentti "Perhe kävelee neljänä". Tan kuvaa sitä tällä tavoin: ”Oireyhtymän geneettinen luonne viittaa käänteiseen vaiheeseen ihmisen evoluutiossa, todennäköisesti johtuen geneettisestä mutaatiosta, käänteisprosessista siirtymisessä kvadropedalismista (kävely neljällä raajalla) bipedalismiin (kävely kahdella). Tässä tapauksessa oireyhtymä vastaa epäjatkuvan tasapainon teoriaa.

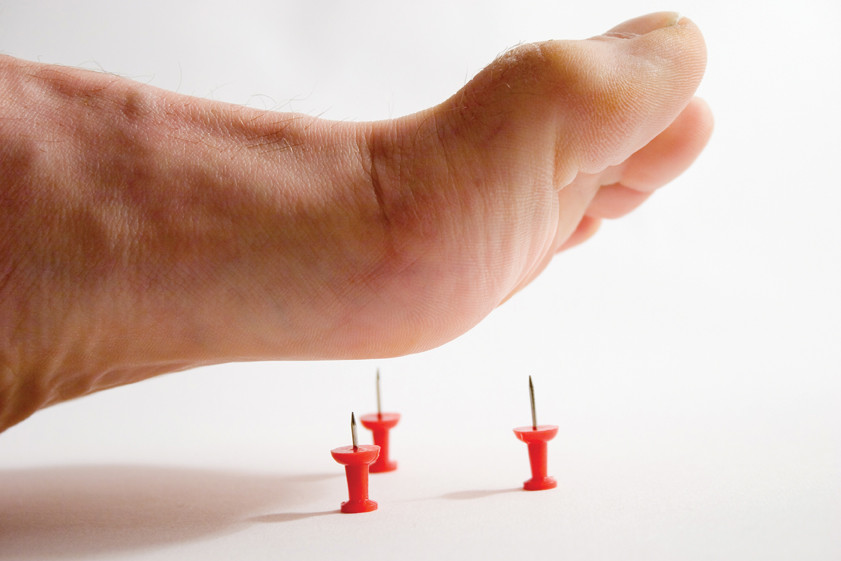
Yksi maailman harvinaisimmista sairauksista: tämäntyyppinen neuropatia diagnosoidaan kahdella miljoonasta ihmisestä. Epänormaalisuus tapahtuu ääreishermoston vaurioista, jotka johtuvat PMP22-geenin ylitarjonnasta. Ensimmäisen tyyppisen perinnöllisen sensorisen neuropatian kehityksen tärkein merkki on käsivarsien ja jalkojen herkkyyden menetys. Ihminen lakkaa käyttämästä kipua ja tuntea lämpötilan muutosta, mikä voi johtaa kudosnekroosiin, esimerkiksi jos murtumaa tai muuta vammaa ei tunnisteta ajoissa. Kipu on yksi kehon reaktioista, mikä ilmoittaa kaikista ”toimintahäiriöistä”, joten kivunherkkyyden menetys on vaarallisten sairauksien havaitsemisen liian myöhäistä, olipa kyse sitten infektioista tai haavaumista.
Tästä epätavallisesta vaivasta kärsivät ihmiset näyttävät olevan paljon ikääntyneempiä, minkä vuoksi sitä kutsutaan joskus käänteisen Benjamin-napin oireyhtymäksi. Perinnöllisistä geneettisistä mutaatioista johtuen ja toisinaan tiettyjen lääkkeiden käytön seurauksena autoimmuunimekanismit ovat häiriintyneet kehossa, mikä johtaa ihonalaisen rasvavaraston nopeaan menetykseen. Useimmiten kärsivät kasvojen, kaulan, yläraajojen ja rungon rasvakudokset, minkä seurauksena ryppyjä ja taittuu. Toistaiseksi vain 200 progressiivisen lipodystrofian tapausta on vahvistettu, ja pääasiassa se kehittyy naisilla. Lääkärit käyttävät hoidossa insuliinia, kasvojen korotuksia ja kollageeni-injektioita, mutta tämä antaa vain väliaikaisen vaikutuksen.

Hypertrichoosia kutsutaan myös "ihmissusi-oireyhtymäksi" tai "Abramsin oireyhtymäksi". Sitä esiintyy vain yhdellä henkilöstä miljardista, ja vain 50 tapausta keskiajalta lähtien on dokumentoitu. Hypertrichoosista kärsiville ihmisille on ominaista liiallinen määrä hiuksia kasvoissa, korvissa ja harteissa. Tämä johtuu orvaskeden ja dermisen välisten siteiden rikkomisesta kolmen kuukauden ikäisen sikiön muodostuessa hiusrakkuloita. Tuloksena olevan dermisen signaalit ”yleensä” kertovat follikkelia niiden muodon. Follikkelit puolestaan \u200b\u200bmyös osoittavat ihokerroksille, että tällä alueella on jo yksi follikkelia, ja tämä johtaa siihen tosiseikkaan, että rungossa karvat kasvavat suunnilleen samalla etäisyydellä toisistaan. Hypertrichoosin tapauksessa nämä yhteydet katkeavat, mikä johtaa liian tiheiden hiusten muodostumiseen niissä kehon osissa, joissa niiden ei pitäisi olla.

Jos olet koskaan kuullut vuohen pyörtymisestä, tiedät mistä synnynnäinen myotonia näyttää - lihasspasmien vuoksi henkilö näyttää jäätyvän hetkeksi. Synnynnäisen (synnynnäisen) myotonian syy on geneettinen poikkeama: mutaation vuoksi luustolihasten kloorikanavien toiminta on häiriintynyt. Lihaskudos Se osoittautuu sekoittuneeksi, tapahtuu mielivaltaisia \u200b\u200bsupistuksia ja rentoutumista, ja patologia voi vaikuttaa jalkojen, käsivarsien, leukojen ja pallean lihaksiin.

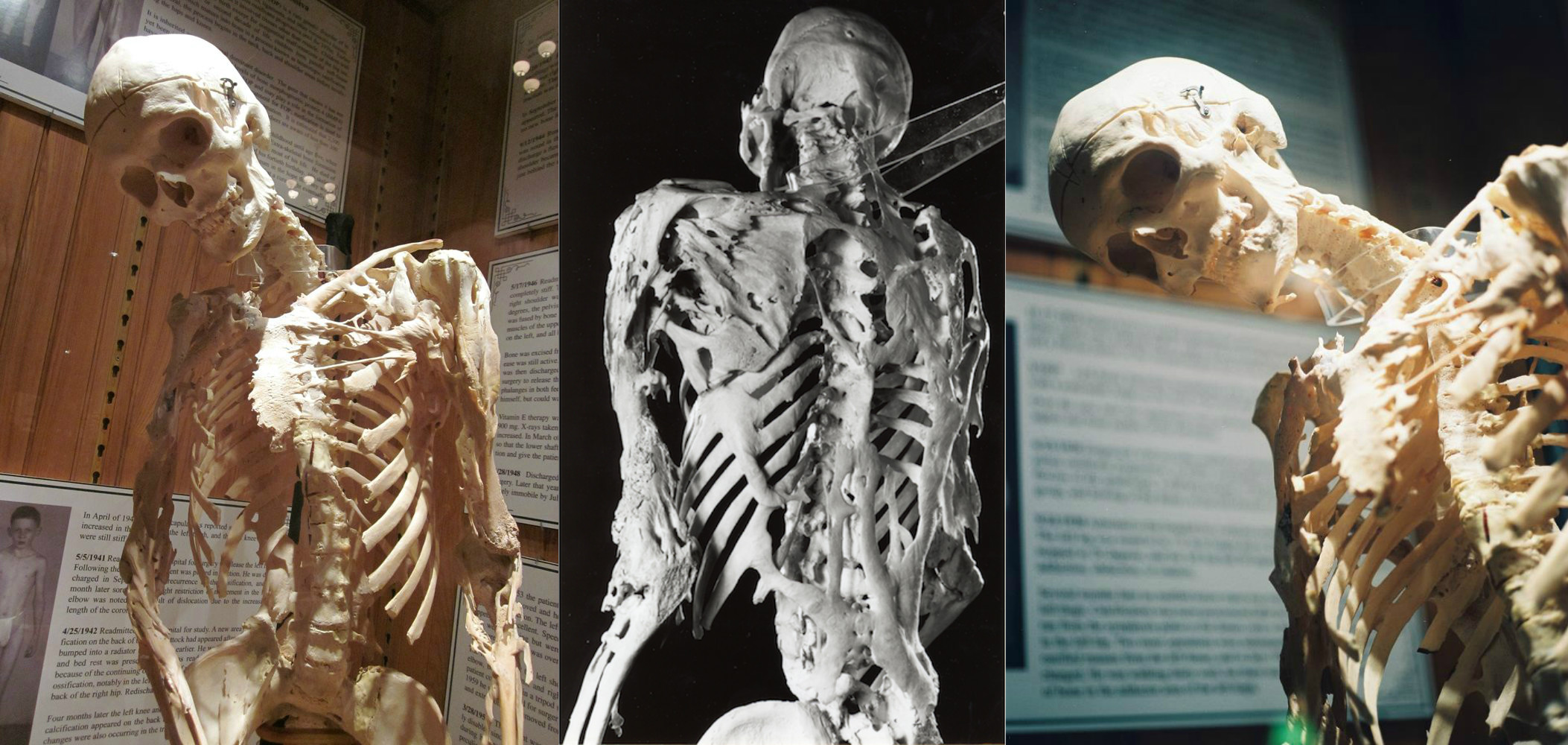
Harvinainen geneettinen sairaus, jossa elimistö alkaa muodostaa uusia luita - luutumista - väärään paikkaan: lihaksen, nivelsiteiden, jänteiden ja muiden sidekudosten sisäpuolelle. Mahdolliset vammat voivat johtaa niiden muodostumiseen: mustelmat, leikkaukset, murtumat, lihaksensisäiset injektiot tai leikkaukset. Tämän vuoksi luutumista ei voida poistaa: leikkauksen jälkeen luu voi vain kasvaa. Fysiologisesti luustot eivät eroa tavallisista luista ja kestävät merkittäviä kuormituksia, mutta ne eivät ole oikeassa paikassa.
FOP tapahtuu ACVR1 / ALK2-geenin mutaation vuoksi, joka koodaa luun morfogeneettistä proteiinireseptoria. Se välittyy henkilölle perinnöllä yhdeltä vanhemmilta, jos hän on myös sairas. On mahdotonta olla tämän taudin kantaja: potilas on joko sairas tai ei. Toistaiseksi FOP on parantumattomien sairauksien joukossa, mutta nyt parhaillaan suoritetaan palovaroteenin nimisen lääkkeen toinen tutkimussarja, jonka avulla voit estää patologiasta vastaavan geenin.
Tämä perinnöllinen ihosairaus ilmenee ihmisen lisääntyneessä herkkyydessä ultraviolettisäteille. Se johtuu altistumisesta johtuvien DNA-vaurioiden korjaamisesta vastaavien proteiinien mutaatiosta. ultravioletti säteily. Ensimmäiset oireet ilmenevät yleensä varhaislapsuudessa (enintään 3 vuotta): kun lapsi on auringossa, hän kärsii vakavista palovammoista vain muutaman minuutin altistumisen jälkeen auringonvalolle. Taudille on ominaista myös pisamien esiintyminen, kuiva iho ja epätasainen muutos ihon värissä. Tilastojen mukaan xeroderma pigmentosa -tartunnan sairastavilla on enemmän syöpää kuin muilla: puuttuessa asianmukaisista ennaltaehkäisevistä toimenpiteistä noin puolelta xerodermasta kärsivistä lapsista kehittyy yksi tai toinen syöpäsairaus kymmeneen ikään asti. Tätä sairautta on kahdeksan tyyppiä, joiden vakavuus ja oireet vaihtelevat. Eurooppalaisten ja amerikkalaisten lääkäreiden mukaan tauti esiintyy noin neljällä miljoonasta ihmisestä.

Utelias nimi sairaudelle, eikö niin? Tälle ”kipeälle” on kuitenkin olemassa tieteellinen termi - desquamative glossitis. Maantieteellinen kieli esiintyy noin 2,58%: lla ihmisistä, ja useimmiten taudilla on kroonisia ominaisuuksia ja pahenee syömisen jälkeen, stressin tai hormonaalisen stressin aikana. Oireet ilmenevät värjäytyneiden saaria muistuttavien sileiden pisteiden kielellä, koska sairaus on saanut niin epätavallisen lempinimen, ja ajan myötä jotkut ”saaret” muuttavat muotoaan ja sijaintiaan riippuen siitä, mikä kielen makuhermoista paranee, ja jotka päinvastoin ovat ärsyttäviä.
Maantieteellinen kieli on käytännössä vaaratonta, jos et ota huomioon lisääntynyttä herkkyyttä mausteisille ruuille tai sen aiheuttamia epämukavuuksia. Lääketiede ei tiedä tämän taudin syitä, mutta on olemassa todisteita geneettisestä taipumuksesta sen kehitykseen.
Paitsi ulkoiset piirteet ja luonnepiirteet, myös joukko terveysongelmia voidaan välittää lapselle hänen biologisista vanhemmistaan.
Perinnölliset sairaudet ovat harvinaisia, mutta yleensä nämä ovat melko vakavia sairauksia, joita ei käytännössä voida hoitaa.
Jokainen ihmiskehon geeni sisältää ainutlaatuisen DNA: n, sillä on oma erityinen koodinsa tietylle ominaisuudelle.
Tässä tilanteessa sinun on hakeuduttava lääkärin apuun - genetiikka, suoritettava geneettinen neuvonta selvittääksesi tietyn geneettisen sairauden riskiasteen
Downin tauti
Nykyään yksi yleisimmistä sairauksista

peritty, on Downin tauti. Tilastot osoittavat, että tällainen sairaus esiintyy yhdellä vastasyntyneellä seitsemästä sadasta vauvasta. Tämän diagnoosin määrittää pääsääntöisesti äitiysaseman erikoislääkäri 3-5 päivän ajan vastasyntyneen lapsen elämästä.
Tämän diagnoosin vahvistamiseksi suoritetaan toimenpide, kuten kariotyypin tutkimus. Se koostuu vastasyntyneen vauvan kromosomisarjan tutkimuksesta. Sairaalla lapsella on seitsemän kromosomia, mikä on yksi enemmän kuin terveellä henkilöllä. Tällainen sairaus esiintyy sekä pojilla että tytöillä. Sukupuoleella ei tässä tapauksessa ole merkitystä.
Sherchevsky-Turner -tauti
Tämä tauti on ominainen vain naislapsille. Ensimmäiset merkit tästä geneettisestä patologiasta löytyvät 10–12-vuotiaina.
Pääpään takana hiukset kasvavat yleensä hitaasti, lisäksi niillä on syvä juuri. Tytöt ovat poissa 15-16-vuotiaana tai jopa vanhempana. Tämä on juuri syy kääntyä asiantuntijan puoleen. Iän myötä tauti voi aiheuttaa ongelmia lapsen henkisessä kehityksessä. Shershevsky-Turner-taudin geneettiselle rakenteelle tytöille on tunnusomaista yhden X-kromosomin puuttuminen.
Klinefelterin tauti
Klinefelterin tauti on geneettinen patologia, jota esiintyy vain pojilla. Ensimmäiset taudin merkit voidaan havaita lapsen ollessa 15-16-vuotias.
Ensimmäiset merkit:

Kun tutkitaan kromosomeja, Klinefelterin taudille on ominaista niiden lisääntynyt lukumäärä: vielä yksi X-kromosomi. Joissakin tapauksissa muita kromosomeja voi olla myös läsnä: U, XX, XU.
Monitekijäiset geneettiset sairaudet
Monitekijäiset geneettiset sairaudet ovat geneettisiä patologioita, joita voi esiintyä vastasyntyneellä missä tahansa perheessä.
Tällöin tällaisten sairauksien kehittymisen syy ei ole pelkästään geneettiset poikkeavuudet, vaan myös joukko vieraita tekijöitä, esimerkiksi huono ekologisuus, vanhempien häiriintynyt rytmi.
Näihin sairauksiin kuuluvat: sepelvaltimo- ja sydänsairaudet, mahalaukun sairaudet sekä verenkiertoelimistön ongelmat.
Syntymävaurioita, jotka liittyvät monitekijäisiin geneettisiin sairauksiin, ovat huulilipu, kitalaki ja spina bifida.
Nykyisin kaikki monet patologiat voidaan havaita nykyaikaisilla laitteilla: Sikiön ultraäänitutkimus voi osoittaa monia poikkeavuuksia lapsen kehityksessä.
Geneettiset sairaudet ovat luonteeltaan harvinaisia \u200b\u200bja monimutkaisia \u200b\u200bsairauksia, joita käytännössä ei voida hoitaa perimän tason häiriöiden vuoksi. Siksi asiantuntijat suosittelevat lapsen suunnittelussa, että ota etukäteen yhteyttä geneetikkoon ongelmien välttämiseksi tulevaisuudessa.
Videon asiantuntijat kertovat geneettisistä sairauksista:
Oletko huomannut virheen? Valitse se ja paina Ctrl + Enterkertoa meille.
Piditkö siitä? Pidä ja pidä sivullasi!
iloinen perhetapahtuma - Onnellisin tapahtuma jokaiselle parille. Vauvan kanssa tapaamisen odottaminen on usein varjostettu ahdistuneiden ajatuksien kanssa hänen terveydestään ja asianmukaisesta kehityksestä. Useimmissa tapauksissa nuorten vanhempien ahdistukset ovat turhia, mutta toisinaan kohtalo kohtelee syntymättömää melko ankarasti: vauva saa äidiltä ja isältä paitsi hiusvärin, silmien muodon ja suloisen hymyn, mutta myös erilaisia \u200b\u200bperinnöllisiä sairauksia.
Lääketieteellisten tilastojen mukaan todennäköisyys synnyttää perinnöllinen patologinen vauva jokaiselle odottaville äideille on 3-5%. Esimerkiksi todennäköisyys lapsille, joilla on Downin oireyhtymä, on 1: 700. Vaikeimmin diagnosoitavissa ja jatkohoitoon reagoitavat ovat harvinaiset, ns. Orvotaudit: epätäydellinen osteogeneesi, epidermolyysi bullosa, Menkesin oireyhtymä, progeria ja monet muut. Nämä geneettisesti perinnölliset sairaudet ovat yleensä uhka lapsen elämälle, lyhentävät merkittävästi lapsen kestoa ja laatua ja johtavat vammaisuuden alkamiseen. Maassamme "harvinaista" pidetään taudina, jonka esiintymistiheys on 1: 10000.
Perinnöllisten sairauksien syyt
 Jokaisella ihmiskehon solulla on erityinen koodi, joka sisältyy kromosomeihin. Kaikkiaan henkilöä on 46 heistä: 22 heistä on autosomaalisia pareja, ja 23. kromosomipari vastaa henkilön sukupuolesta. Kromosomit puolestaan \u200b\u200bkoostuvat monista geeneistä, jotka kuljettavat tietoa kehon tietystä ominaisuudesta. Aivan ensimmäinen hedelmöittymisen yhteydessä muodostunut solu sisältää 23 äidin kromosomia ja saman määrän isäisiä. Geenin tai kromosomin virhe johtaa geneettisiin poikkeavuuksiin.
Jokaisella ihmiskehon solulla on erityinen koodi, joka sisältyy kromosomeihin. Kaikkiaan henkilöä on 46 heistä: 22 heistä on autosomaalisia pareja, ja 23. kromosomipari vastaa henkilön sukupuolesta. Kromosomit puolestaan \u200b\u200bkoostuvat monista geeneistä, jotka kuljettavat tietoa kehon tietystä ominaisuudesta. Aivan ensimmäinen hedelmöittymisen yhteydessä muodostunut solu sisältää 23 äidin kromosomia ja saman määrän isäisiä. Geenin tai kromosomin virhe johtaa geneettisiin poikkeavuuksiin.
Geneettisiä häiriöitä on erityyppisiä: yksi geenivika, kromosomivika ja monimutkainen vika.
Yhden geenivikavoidaan välittää yhdeltä tai molemmilta vanhemmilta. Lisäksi, koska äiti ja isä ovat recessiivisen geenin kantajana, eivät ehkä edes tiedä sairaudestaan. Tällaisia \u200b\u200bsairauksia ovat progeria, Menkesin oireyhtymä, bulloosinen epidermolyysi, epätäydellinen osteogeneesi. 23-parin kromosomilla välitettyä vikaa kutsutaan X-linkitetyksi. Jokainen henkilö perii X-kromosomin äidiltä, \u200b\u200bmutta isältä hän voi vastaanottaa Y-kromosomin (tässä tapauksessa syntyy poika) tai X-kromosomin (tyttö ilmestyy). Jos pojan X-kromosomissa löydetään viallinen geeni, sitä ei voida tasapainottaa toisella terveellä X-kromosomilla, ja siksi patologia todennäköisesti kehittyy. Tämä vika voi tarttua taudin äidistä tai muodostaa täysin arvaamattomasti.
Kromosomivika - niiden rakenteen ja lukumäärän muutos. Periaatteessa sellaiset viat muodostuvat vanhempien munien ja siemennesteen muodostumisen aikana. Alkiossa tapahtuu kromosomaalinen vika, kun nämä solut sulautuvat. Tällainen patologia ilmenee pääsääntöisesti vakavien häiriöiden muodossa fyysisessä ja henkisessä kehityksessä.
Monimutkaiset viat syntyvät ympäristötekijöiden geenille tai geeniryhmälle altistumisen seurauksena. Näiden sairauksien leviämismekanismia ei vieläkään ymmärretä täysin. Lääkäreiden oletusten mukaan lapsi perii vanhemmalta erityisen herkkyyden tietyille ympäristötekijöille, joiden vaikutuksesta tauti voi lopulta kehittyä.
Diagnoosi synnytysajalla
 Lasten perinnölliset sairaudet voidaan havaita jopa synnytyksen aikana. Joten äskettäin, monissa neuvotteluissa, kaikille naisille suoritetaan testi, joka määrittelee AFP: n, estrogeenin ja hCG: n hormonitasot 18 raskausviikkoa . Se auttaa määrittämään lapsen kehityksen patologian, joka johtuu kromosomivirheistä. On syytä huomata, että tämän seulonnan avulla voit tunnistaa vain osan geneettisistä häiriöistä, kun taas nykyinen perinnöllisten sairauksien luokittelu on monimutkainen järjestelmä, joka sisältää noin kaksi tuhatta sairautta, tilaa ja oireyhtymää.
Lasten perinnölliset sairaudet voidaan havaita jopa synnytyksen aikana. Joten äskettäin, monissa neuvotteluissa, kaikille naisille suoritetaan testi, joka määrittelee AFP: n, estrogeenin ja hCG: n hormonitasot 18 raskausviikkoa . Se auttaa määrittämään lapsen kehityksen patologian, joka johtuu kromosomivirheistä. On syytä huomata, että tämän seulonnan avulla voit tunnistaa vain osan geneettisistä häiriöistä, kun taas nykyinen perinnöllisten sairauksien luokittelu on monimutkainen järjestelmä, joka sisältää noin kaksi tuhatta sairautta, tilaa ja oireyhtymää.
Tulevien vanhempien tulisi pitää mielessä, että analyysin tulosten perusteella tiettyä sairautta ei diagnosoida, vaan määritetään vain sen todennäköisyys ja päätetään lisätutkimusten tarpeesta.
lapsivesitutkimus- toimenpide, jonka aikana lääkäri ottaa ohutta ja pitkää neulaa käyttämällä amnioottista nestenäytettä ja tunkeutuu naisen kohtuun vatsan läpi. Aikaisemmin nainen lähetetään ultraäänitutkimukseen sikiön sijainnin määrittämiseksi ja paras paikka neulan viemiseksi. Joskus ultraääni suoritetaan suoraan amniokentesis-toimenpiteen aikana.
Tämän tutkimuksen avulla voit tunnistaa monet kromosomaalivirheet, määrittää lapsen keuhkojen kehitysasteen (tarvittaessa toimitus ennen suunniteltua päivämäärää), määrittää tarkasti lapsen sukupuoli (jos kyseessä on tiettyyn sukupuoleen liittyvien sairauksien uhka). Tuloksena olevan nesteen tutkimus kestää useita viikkoja. Tämän menettelyn haittana on, että se voidaan suorittaa raskauden aikana pidemmälle 16 viikkoa , mikä tarkoittaa, että naisella on hyvin vähän aikaa tehdä päätös raskauden lopettamisesta. Lisäksi toisin kuin ensimmäisellä kolmanneksella, abortti niin pitkään on erittäin vaarallinen toimenpide sekä naisen fyysiselle että henkiselle terveydelle. Spontaanin abortin riski tämän tutkimuksen jälkeen on 0,5-1%.
Korionin (sikiötä ympäröivän kudoksen raskauden varhaisessa vaiheessa) tutkimuksen avulla on myös mahdollista tunnistaa sikiön geneettiset häiriöt, mukaan lukien melko harvinaisten sairauksien, kuten bulloosisen epidermolyysin, diagnoosi, epätäydellinen osteogeneesi. Tämän toimenpiteen aikana lääkäri työntää ohut putken emättimen läpi naisen kohtuun. Koorionin palaset imetään putken läpi ja lähetetään sitten analysoitavaksi. Tämä toimenpide on kivuton, ja se voidaan suorittaa jo 9 viikkoa raskauden aikana, tutkimuksen tulokset ovat valmiita yhden tai kahden päivän sisällä. Huolimatta ilmeisistä eduista, tämä toimenpide ei ole kovin kysytty spontaanin abortin (2-3%) ja erilaisten raskauden häiriöiden suuren riskin vuoksi.
Koorion ja amniosenteesi tutkimuksen indikaatiot ovat:
- tulevan äidin ikä on yli 35 vuotta;
- kromosomaaliset viat yhdessä tai molemmissa vanhemmissa;
- lapsen avioparissa syntyminen, jolla on kromosomivirheitä;
- tulevat äidit, joiden perheissä oli X-linkitettyjä sairauksia.
Jos tutkimukset ovat vahvistaneet geneettisen häiriön esiintymisen, vanhempien on punnittuna kaikki edut ja haitat on tehtävä elämässään vaikein valinta: pitää raskaus yllä tai lopettaa se, koska perinnöllisten sairauksien hoito tässä vaiheessa on valitettavasti mahdotonta.
Diagnostiikka vauvan syntymän jälkeen
 Harvinaisten perinnöllisten sairauksien diagnosointi voi perustua laboratoriokokeisiin. Jo usean vuoden ajan kaikissa synnytyssairaaloissa, viidentenä päivänä vauvan syntymän jälkeen, seulotaan vastasyntyneet, joiden aikana diagnosoidaan joukko harvinaisia \u200b\u200bperinnöllisiä sairauksia: fenyyliketonuria, kilpirauhasen vajaatoiminta, kystinen fibroosi, galaktosemia ja adrenogenitaalinen oireyhtymä.
Harvinaisten perinnöllisten sairauksien diagnosointi voi perustua laboratoriokokeisiin. Jo usean vuoden ajan kaikissa synnytyssairaaloissa, viidentenä päivänä vauvan syntymän jälkeen, seulotaan vastasyntyneet, joiden aikana diagnosoidaan joukko harvinaisia \u200b\u200bperinnöllisiä sairauksia: fenyyliketonuria, kilpirauhasen vajaatoiminta, kystinen fibroosi, galaktosemia ja adrenogenitaalinen oireyhtymä.
Muut sairaudet diagnosoidaan oireiden ja oireiden perusteella, joita voi esiintyä sekä vastasyntyneillä että monien vuosien ajan syntymän jälkeen. Bulloosisen epidermolyysin ja epätäydellisen osteogeneesin oireet ilmenevät useimmissa tapauksissa heti syntymän jälkeen, ja progeriat diagnoositaan useimmiten vasta 2-3 vuoden ikäisenä lapsen elämästä.
Tavallisella lastenlääkärillä on erittäin vaikea tunnistaa harvinaisia \u200b\u200bsairauksia, lääkäri ei yksinkertaisesti huomaa heidän oireitaan rutiininomaisen tapaamisen aikana. Siksi äidin on oltava erityisen tarkkaavainen omaan lapsiinsa ja kiinnitettävä huomiota uhkaaviin oireisiin: ei ikään liittyviin motorisiin taitoihin, kouristuksiin, riittämättömään painonnousuun, epäluonnollisiin väreihin ja ulosteen hajuun. Huolenaiheen tulisi olla lapsen kasvuprosessin voimakas lisääntyminen tai hidastuminen. Tämä voi viitata sellaisen sairauden esiintymiseen kuin kääpiö. Jos tällaisia \u200b\u200boireita ilmenee, vanhempien on aina otettava yhteys lääkäriin ja vaadittava lapsen perusteellista tutkimista, koska perinnöllisten sairauksien oikea-aikainen diagnosointi ja oikean hoito-ohjelman valinta voivat auttaa pitämään vauvan terveyttä ja joskus elämää.
Kuinka geneettiset sairaudet hoidetaan?
Vaikka useimpia perinnöllisiä sairauksia ei voida hoitaa, moderni lääketiede pystyy parantamaan huomattavasti sairaiden lasten elinajanodotetta ja parantamaan sen laatua. Nykyään tällaiset sairaudet eivät ole lause, vaan pikemminkin elämäntapa, joka antaa lapselle mahdollisuuden kehittyä normaalisti edellyttäen, että hän saa tarvittavaa hoitoa: lääkkeiden ottamista, voimistelua, erityisruokavalioita. Lisäksi mitä aikaisemmin on mahdollista diagnosoida, sitä menestyvämpi on perinnöllisten sairauksien hoito.
Viime aikoina synnytyksen (synnytyksen) hoitomenetelmiä käytetään yhä enemmän: lääkkeiden ja jopa kirurgisten toimenpiteiden avulla.
Lasten tauti on vaikea testi koko perheelle. Näissä olosuhteissa vanhempien kannalta on erittäin tärkeää sukulaisten tuki ja viestintä muiden äitien ja isien kanssa, jotka ovat samanlaisessa tilanteessa. Erilaiset vanhempien perheet, joissa on lapsia, joilla on harvinaisia \u200b\u200bgeneettisiä sairauksia, tarjoavat suurta apua näille perheille.
Kuinka estää perinnöllisiä sairauksia?
Oikea raskaussuunnittelu, jonka pääpaino on perinnöllisten sairauksien ehkäisemisessä, auttaa välttämään sairaan lapsen syntymää. Riskialttiiden vanhempien tulisi ehdottomasti käydä geneetikolla:
- vanhempien ikä on vähintään 35 vuotta;
- yhden tai useamman lapsen läsnäolo, jolla on perinnöllinen sairaus;
- puolisoiden tai heidän lähisukulaistensa harvinaiset sairaudet;
- parit, jotka ovat huolissaan terveen vauvan saamisesta.
Geneettinen konsultti, joka perustuu lääketieteellisestä tutkimuksesta saatuihin tietoihin, sekä tietoihin perheen historiasta, sukulaisten sairauksista, abortista ja keskenmenoista, laskee todennäköisyyden saada lapsi, jolla on geneettinen sairaus. Tapahtuu, että pari, jolla on hyvät mahdollisuudet synnyttää sairas lapsi, luopuu suunnitelmista tässä liittoutumassa ja hankkii muiden kumppaneiden kanssa täysin terveitä lapsia.
Tytöt! Tehdään postitukset.
Tämän ansiosta asiantuntijat ottavat meihin yhteyttä ja vastaavat kysymyksiimme!
Voit myös esittää kysymyksesi alla. Sinun tai asiantuntijoiden kaltainen antaa vastauksen.
Kiitos ;-)
Kaikki terveet lapset!
Ps. Tämä koskee myös poikia! Täällä on yksinkertaisesti enemmän tyttöjä ;-)
Pidätkö tavaroista? Tuki - repost! Yritämme puolestasi ;-)