ЭТО ПЕРЕПОСТ! Оригинал взят у eka_tyryshkina в Ошибки природы: люди с редкими заболеваниями.
Заболела я тут на днях, как всегда, как нам только куда надо идти в гости, я болею! Толи моя болезнь реагирует на плановость мероприятия, толи еще на что то, но хорошо что не реагирует на работу. Вообщем болезнь у меня не простая)
И вот болея дома в поздний час, уже переделав все дела, перечитав и пролистав все интресные сайты, вдруг не ожиданно для себя решила узнать о самых редких болязнях на планете и знаете, столько интересного и шокируешего!!!
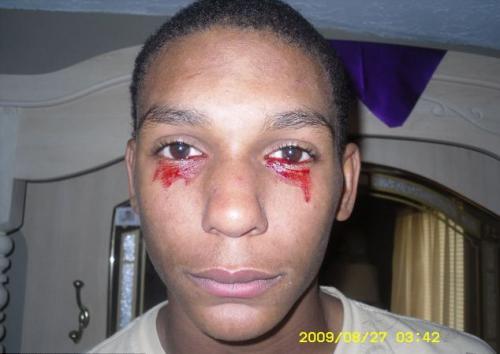
Гемолакрия
(«кровавые слезы») наблюдается у одного человека на миллион.Кровь, вместо слезной жидкости, начинает течь из глаз внезапно, и это может продолжаться около часа. За день больной обливается кровавыми слезами от 3 до 20 раз.
Точная причина этого заболевания до конца не изучена, а стало быть и лечению не поддаётся. Медицинские специалисты пока выдвигают версии что гемолакрия это одно из заболеваний крови или опухоли.
На фото - 15-летний Кальвино Инмэн (штат Теннеси, США)

Синдром Вампира
С диагнозом «синдром вампира»
(эктодермальная дисплазия) в мире насчитывается всего 7 тысяч человек.
Помимо мертвенно-бледной кожи и острых клыков (при отсутствии части зубов), у больных редкие и тонкие волосы, способность потеть снижена, поэтому их организм подвержен перегреву. Симптомы проявляются в детстве, однако выявить заболевание можно уже на стадии беременности с помощью генетических тестов.
Мальчики вынуждены носить темные очки и пользоваться солнцезащитным кремом, когда выходят на улицу, поскольку они не могут находиться под прямыми солнечными лучами.
При этом физическое развитие и двигательная активность остается в норме.
Сама болезнь неизлечима, коррекции поддаются только симптомы. В частности, можно восстановить нормальную форму зубов.
Болезнь Саймона была диагностирована в младенчестве. Когда Мэнди была беременна второй раз, ее предупредили, что у второго ребенка может быть такое же заболевание.
Однако Саймон рос и развивался хорошо, поэтому родители пошли на этот риск.
Мальчики говорят: "некоторые дети смеются над нашей внешностью, но наши друзья думают, что это круто"
На фото - Саймон (13 лет) и Джордж (11 лет) Каллен (Саффолк, Великобритания).

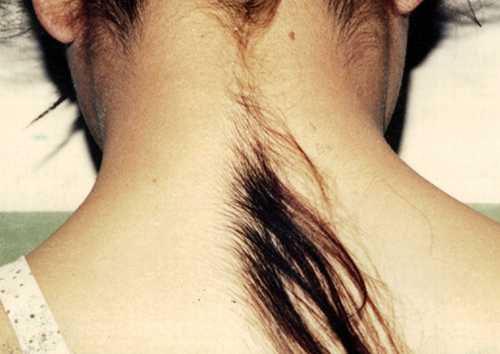
Гипертрихоз
(«синдром оборотня») - это заболевание, проявляющееся в избыточном росте волос, не свойственному данному участку кожи, не соответствующему полу и возрасту.Во всем мире зарегистрировано всего чуть более сорока таких пациентов, так что самый удобный для них способ заработать - демонстрировать свое уродство...Они подают заявки в книгу рекордов Гиннеса,- чтобы прославится и заработать деньги... Китайцу Юй Чжэньхуан это удалось на все сто - благодаря своей сверхволосатости он основал популярнейшую в своей стране рок-группу и стал миллионером.
Неизвестно, отчего происходит подобная мутация. И лечение от гипертрихоза тоже пока еще никто не разработал. Косметологи умеют только удалять волосы на достаточно долгий срок...
На фото - 6-летняя Нат Сасуфан
(Таиланд), 2007 год

На фото - 33-летний Юй Чжэньхуан (Китай), самый волосатый человек в мире

Слоновая болезнь
(«синдром Протея», слоновость, элефантиаз, элефантиазис) - увеличение размеров какой-либо части тела за счёт болезненного разрастания кожи и подкожной клетчатки.Всего в мире насчитывается примерно 120 человек с этим неизлечимым заболеванием…
А самым известным больным был «человек-слон» - Джозеф Меррик. О знаменитом британце в 1980 году режиссёр Дэвид Линч даже снял фильм, который был выдвинут на «Оскар» по восьми номинациям… Речь в фильме шла о человеческом достоинстве… Грим Джона Херта, который и сыграл Меррика, был создан на основе представленного в Королевской больнице Лондона заспиртованного тела Джозефа Меррика. Его накладка ежедневно занимала у актёра 12 часов в день…
На фото - 35-летняя Менди Селларс
(Великобритания)

Генную аномалию, заключающуюся в ускоренном старении организма
,
- прогерию
- подразделяют на детскую (синдром Гетчинсона) и взрослую (синдром Вернера). Впервые о синдроме преждевременного старения заговорили 100 лет назад. И не удивительно, такие случаи встречаются один раз на 4-8 миллионов младенцев. Прогерия (от греческого prо - раньше, gerontos - старец) - крайне редкое генетическое заболевание, ускоряющее процесс старения примерно в 8-10 раз. Проще говоря, ребенок за один год стареет на 10-15 лет. Восьмилетний выглядит на 80 лет - с сухой морщинистой кожей, облысевшей головой... Эти дети обычно погибают в 13-14 лет после нескольких инфарктов и инсультов на фоне прогрессирующего атеросклероза, катаракты, глаукомы, полной потери зубов и т.д. И лишь немногие живут до 20 лет или дольше.
Сейчас в мире известны всего 42 случая заболевания людей прогерией... Из них 14 человек проживают на территории Соединенных Штатов, 5 - в Росии, остальные в Европе...
В настоящее время существует несколько организаций, оказывающих помощь маленьким старичкам и их семьям. В Интернете есть сайты, посвященные именно этой проблеме, некоторые из них открыты медиками или социальными работниками, другие - семьями больных.
На фото - 24-летний Леон Бот



38-летний человек-дерево
Деде Косвара, проживающий на острове Ява, в Индонезии, стал знаменитым на весь мир из-за вируса папилломы человека, который обычно приводит к появлению небольших бородавок, но в случае с индонезйцем до неузнаваемости деформировал его конечности.
Проблема Дедэ состояла в том, что он имел редкое генетическое отклонение, которое не позволяло его иммунной системе сдержать рост этих бородавок. Поэтому вирус смог "завладеть клеточным механизмом его клеток кожи", отдавая им приказы вырабатывать большое количество рогового вещества, из которого и состояли. У Дедэ обнаружилось также низкое содержание лейкоцитов в крови.


Болезнь Бабочки
Буллёзный эпидермолиз в гиперпластической форме — это генетическое заболевание, которое проявляется в первые дни жизни. По сути, кожа новорождённого настолько нежная, что любое прикосновение приводит к возникновению ран и пузырей. Больше всего страдают выступающие участки: локти, колени, стопы, руки. Возникшая язва, с которой слоями сходит кожа, долго не заживает, из неё выделяется жидкость. После образуется большой малиновый шрам.
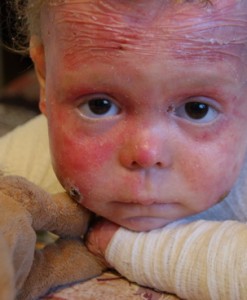
Лечения данного заболевания нет, возможно только облегчение симптомов. Не так давно на всю Россию прогремела история Лизы Кунигель, которая живёт с буллёзным эпидермолизом уже почти десять лет. Несколько раз в день ей необходимы перевязки и обработка противомикробными мазями и гелями. Кроме этого, все 9 лет Лизу сопровождает боль.
Синдром русалки
Одной из редчайших аномалий в развитии является сиреномелия, в народе называемая «синдромом русалки». При данном дефекте новорожденные появляются на свет со срощенными ногами, похожими на рыбий хвост. У них функционирует только одна почка, отсутствуют гениталии. Из-за обширного поражения внутренних органов такие младенцы обычно вскоре умирают. Болезнь встречается у одного из 100 000 новорожденных. За все годы наблюдений лишь трое малышей смогли выжить. Одной из них была Шайло Пепин.
Шайло родилась в 1999 году и стала самым знаменитым ребенком с «синдромом русалки». За те 10 лет, что она смогла прожить у нее появились тысячи друзей по всему миру, которые поддерживали девочку и её маму. Шайло старалась вести полноценную жизнь - она, как и все обычные дети ходила в школу, посещала занятия танцев, ездила в парки развлечений. Известной девочка стала после участия в шоу Опры Уинфри. Learning Chanel снял о ней несколько фильмов, ей посвящены сотни сайтов в интернете.

История с Шайло - удивительная история о чуде. Ребенок, который все свое детство боролся за то, что выжить. Маленькая девочка, умевшая радоваться каждому дню, несмотря на неизлечимую болезнь.
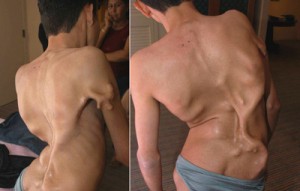
Болезнь Мюнхеймера
Фибродисплазия - заболевание крайне редкое. Официальная статистика такова: 1 больной на 2 000 000 человек. Болезнь Мюнхеймера возникает в результате мутации гена и при рождении проявляется во внешних дефектах. У младенца искривлены большие пальцы стоп, позвоночник. Патология приводит к инвалидности, ранней смертности. Там где должны проходить противовоспалительные процессы, начинает образовываться костный нарост, поэтому часто заболевание называют «болезнью второго скелета».
Любой, даже незначительный ушиб, может привести к развитию остекленения на пораженном месте.На сегодняшний момент официального лечения от смертельной болезни не существует. Ученые разработали препарат, который теоретически может бороться с недугом. Однако, необходимых клинических исследований проведено ещё не было. Увы, провести их очень трудно - во всем мире насчитывается не более 600 человек с болезнью Мюнхеймера.
Феномен «Линий Блашко» характеризуется наличием странных полос по всему телу. Линии Блашко - это невидимый рисунок, заложенный в ДНК. И проявлением заболевания становится видимость этого рисунка.
Обычно рисунок на спине имеет V-образную форму, а на груди, животе и на боках - S-образную.
Причиной заболевания может являться мозаицизм. В любом случае, появление линий Блашко никак не связано с нервной, мускульной и лимфатической системами человека.
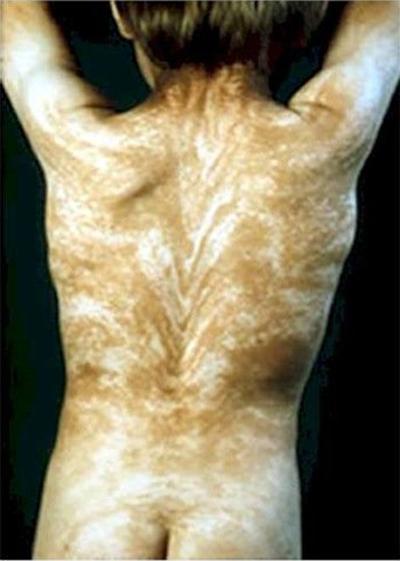
Еще одно аномальное заболевание - акантокератодермия , или «синдром синей кожи». Люди с таким диагнозом могут иметь голубую, цвета индиго, сливовую или практически фиолетовую кожу.В 60-х годах прошлого века в штате Кентукки проживало целое семейство «синих» людей. Они были известны как Синие Фьюгейты. Эта особенность передавалась из поколения в поколение.


Редкими заболеваниями страдает около 6% жителей Земли, и это число продолжает увеличиваться. Все уникальные болезни имеют различную природу, однако подавляющее большинство феноменальных недугов связано с генетическими аномалиями и инфекциями.
Генетические заболевания уникальны тем, что не зависят от образа жизни человека, от них нельзя застраховаться просто перестав есть жирную пищу или начав делать зарядку по утрам. Они возникают в результате мутации и могут передаваться из поколения в поколение.
Редчайшая наследственная болезнь, при которой человек погибает от неспособности заснуть. До сих пор она отмечалась лишь в 40 семьях по всему миру. Фатальная бессонница обычно проявляется между 30 и 60 годами (чаще всего - после 50 лет) и продолжается от 7 до 36 месяцев. По мере того, как заболевание прогрессирует, пациент страдает от всё более тяжелых нарушений сна, причем никакие снотворные ему не помогают. На первой стадии бессонница сопровождается паническими атаками и фобиями, на второй к ним прибавляются галлюцинации и повышенное потоотделение. На третьей стадии болезни человек полностью теряет способность спать и начинает выглядеть намного старше своих лет. Затем развивается деменция, и пациент погибает - как правило, от истощения или пневмонии.

Синдром нарколепсии-катаплексии, для которого характерны внезапные приступы сна и расслабления мускулатуры тела, тоже имеет генетическую природу и возникает из–за нарушений быстрой фазы сна. Он встречается намного чаще фатальной семейной бессонницы: у 40 из каждых 100 тыс. человек, в равной степени у мужчин и у женщин. Человек, страдающий нарколепсией, способен внезапно заснуть на несколько минут посреди дня. «Сонные атаки» напоминают фазу быстрого сна и могут случаться очень часто: до 100 раз в день, с предшествующей им головной болью, либо без неё. Они часто провоцируются бездеятельностью, но могут возникать в совершенно неподходящее время: во время полового акта, занятий спортом, вождения. Просыпается человек отдохнувшим.
![]()
Синдром Юнера Тана (СЮТ) характерен прежде всего тем, что люди, страдающие им, ходят на четвереньках. Открыл его турецкий биолог Юнер Тан после изучения пяти членов семьи Улас в сельской местности Турции. Чаще всего люди с СЮТ пользуются примитивной речью и имеют врождённую мозговую недостаточность. В 2006-м году о семье Улас был снят документальный фильм под названием «Семья, ходящая на четвереньках». Тан описывает это так: «Генетическая природа синдрома предполагает обратную ступень в эволюции человека, вызванную, скорее всего, генетической мутацией, обратному процессу перехода от квадропедализма (хождения на четырёх конечностях) к бипедализму (хождению на двух). В этом случае синдром соответствует теории прерывистого равновесия.

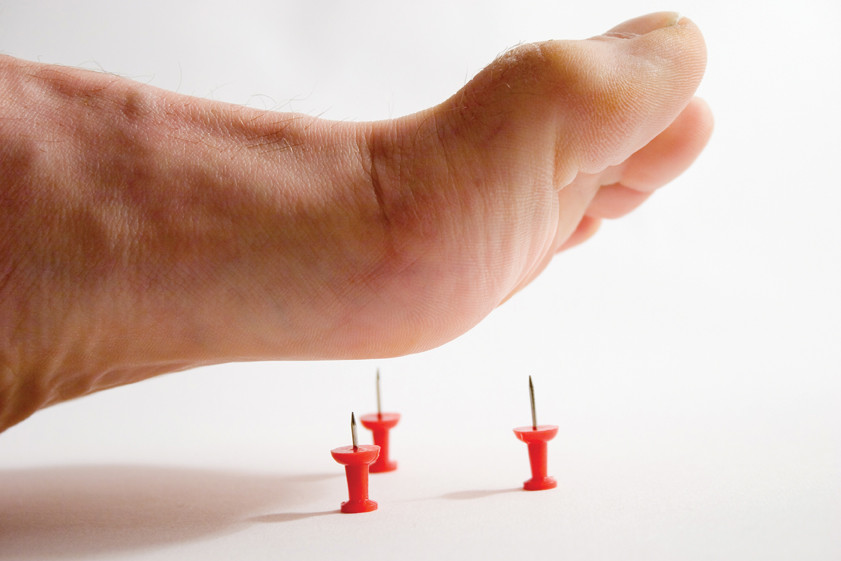
Одно из самых редких в мире заболеваний: этот вид нейропатии диагностируют у двух человек из миллиона. Аномалия возникает из-за поражения периферической нервной системы, возникающего вследствие переизбытка гена PMP22. Главным признаком развития наследственной сенсорной нейропатии первого типа является потеря чувствительности рук и ног. Человек перестаёт испытывать боль и ощущать изменение температуры, что может привести к возникновению некроза тканей, например, если вовремя не распознать перелом или другую травму. Боль - одна из реакций организма, сигнализирующих о каких-либо «неполадках», поэтому потеря болевой чувствительности чревата слишком поздним выявлением опасных заболеваний, будь то инфекции или язвы.
Люди, страдающие этим необычным недугом, выглядят значительно старше своего возраста, поэтому его иногда называют «обратный синдром Бенджамина Баттона». Из-за наследственной генетической мутации, а иногда в результате применения некоторых лекарственных препаратов в организме нарушаются аутоиммунные механизмы, что приводит к быстрой потере подкожных жировых запасов. Чаще всего страдает жировая ткань лица, шеи, верхних конечностей и туловища, вследствие чего возникают морщины и складки. Пока подтверждено лишь 200 случаев прогрессирующей липодистрофии, и главным образом она развивается у женщин. При лечении врачи используют инсулин, «подтяжки» лица и инъекции коллагена, однако это даёт лишь временный эффект.

Гипертрихоз также называют «синдромом оборотня» или «синдромом Абрамса». Он проявляется только у одного человека из миллиарда, и только 50 случаев со времён Средневековья были задокументированы. Люди, страдающие гипертрихозом, отличаются чрезмерным количеством волос на лице, ушах и плечах. Это происходит из-за нарушения связей между эпидермисом и дермой во время формирования у трёхмесячного плода волосяных фолликул. Как правило, сигналы от образующейся дермы «сообщают» фолликулам их форму. Фолликулы тоже, в свою очередь, сигнализируют кожным слоям, что в этой области одна фолликула уже есть, и это приводит к тому, что на теле волоски растут на приблизительно одинаковом расстоянии друг от друга. В случае с гипертрихозом эти связи нарушены, что приводит к образованию слишком плотного волосяного покрова на тех участках тела, где его быть не должно.

Если вы когда-нибудь слышали о козьем обмороке, то примерно знаете, как выглядит конгенитальная миотония - из-за мышечных спазмов человек на некоторое время будто замирает. Причиной возникновения конгенитальной (врождённой) миотонии является генетическое отклонение: вследствие мутации нарушается работа хлорных каналов скелетных мышц. Мышечная ткань оказывается «сбитой с толку», возникают произвольные сокращения и расслабления, причём патология может затрагивать мускулатуру ног, рук, челюстей и диафрагмы.

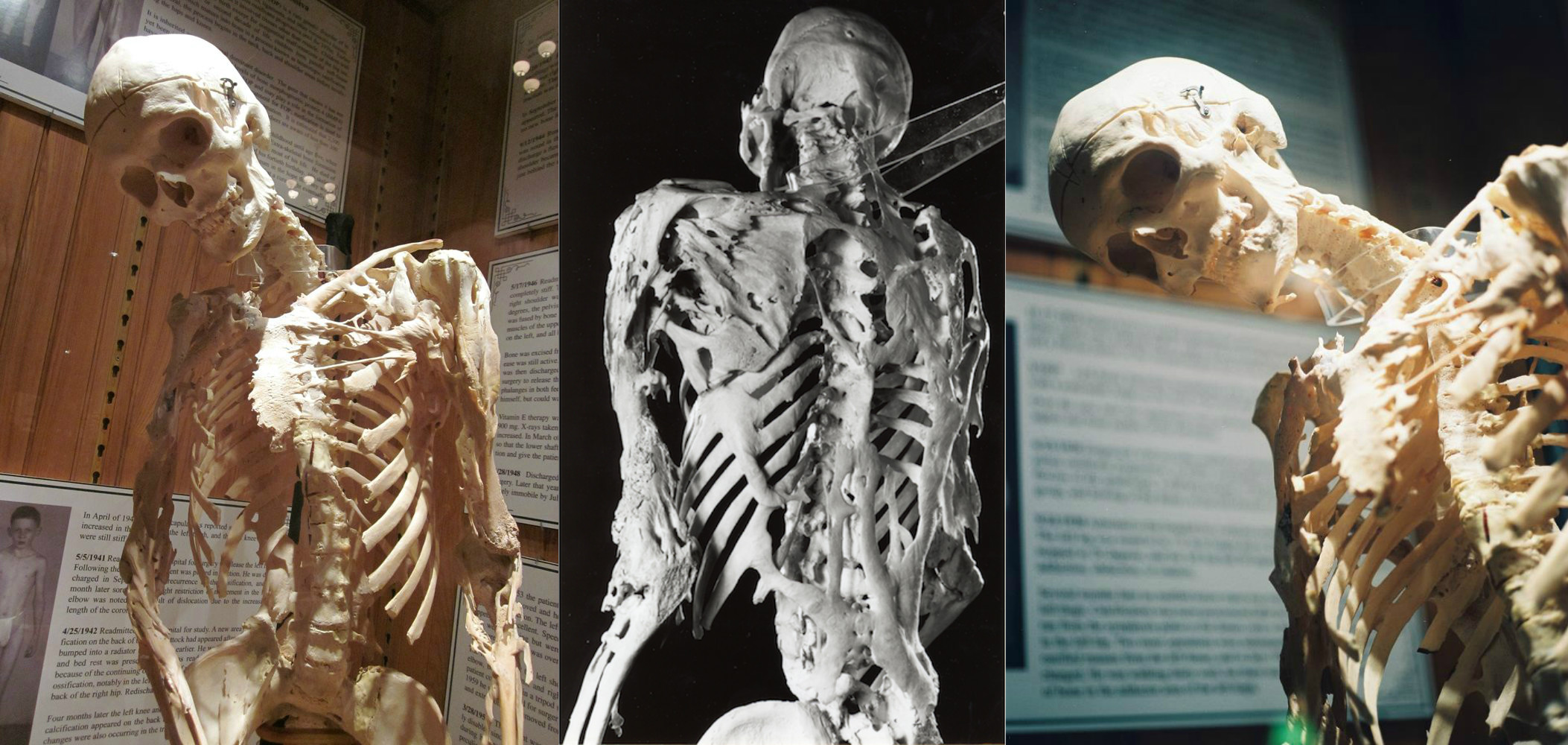
Редкое генетическое заболевание, при котором организм начинает формировать новые кости - оссификаты - в неположенных местах: внутри мышц, связок, сухожилий и других соединительных тканей. К их образованию может привести любая травма: ушиб, порез, перелом, внутримышечная инъекция или операция. Из–за этого удалять оссификаты нельзя: после хирургического вмешательства кость может только сильнее разрастись. Физиологически оссификаты не отличаются от обыкновенных костей и могут выдерживать значительные нагрузки, вот только находятся не там, где надо.
ФОП возникает из–за мутации в гене ACVR1/ALK2, кодирующем рецептор костного морфогенетического белка. Она передается человеку по наследству от одного из родителей, если он тоже болен. Быть носителем этого заболевания нельзя: пациент либо болен, либо нет. Пока ФОП относится к числу неизлечимых болезней, однако сейчас проводится вторая серия испытаний препарата под названием паловаротен, который позволяет заблокировать ген, ответственный за патологию.
Это наследственное заболевание кожи проявляется в повышенной чувствительности человека к ультрафиолетовым лучам. Возникает оно из-за мутации белков, ответственных за исправление повреждений ДНК, появляющихся при воздействии ультрафиолетового излучения. Первые симптомы обычно проявляются в раннем детстве (до 3-х лет): когда ребёнок находится на солнце, у него возникают серьёзные ожоги уже после нескольких минут воздействия солнечных лучей. Также заболевание характеризуется появлением веснушек, сухостью кожи и неравномерным изменением цвета кожного покрова. Согласно статистике, люди с пигментной ксеродермой более других подвержены риску развития онкологических заболеваний: при отсутствии надлежащих профилактических мер, примерно у половины детей, страдающих ксеродермой, к десяти годам развиваются те или иные раковые заболевания. Существует восемь видов этого недуга различной тяжести и симптомов. По данным европейских и американских медиков, болезнь встречается примерно у четырёх человек из миллиона.

Любопытное название для болезни, не так ли? Впрочем, существует и научный термин для обозначения этой «болячки» - десквамативный глоссит. Географический язык проявляется примерно у 2,58% людей, причём чаще всего заболевание имеет хронические свойства и обостряется после еды, во время стресса или гормональных стрессов. Симптомы проявляются в возникновении на языке обесцвеченных гладких пятен, напоминающих острова, потому заболевание и получило столь необыкновенное прозвище, причём со временем некоторые «острова» меняют свою форму и расположение, в зависимости от того, какие из вкусовых сосочков, расположенных на языке, заживают, а какие, наоборот, раздражаются.
Географический язык практически безвреден, если не брать в расчёт повышенную чувствительность к острой пище или некоторый дискомфорт, который он может причинять. Медицине неизвестны причины возникновения этой болезни, но есть данные о генетической предрасположенности к её развитию.
К ребенку от его биологических родителей могут передаваться не только внешние черты и особенности характера, но и ряд проблем со здоровьем.
Наследственные заболевания встречаются редко, но, как правило, это довольно тяжелые болезни, которые практически не поддаются лечению.
Каждый ген организма человека содержит в себе уникальную ДНК, он имеет свой уникальный код конкретного признака.
В данной ситуации необходимо обратиться за помощью к врачу – генетику, пройти генетическую консультацию, чтобы узнать степень риска возникновения того или иного генетического заболевания
Болезнь Дауна
На сегодняшний день одной из наиболее распространенных болезней, которая

передается по наследству, является Болезнь Дауна. Статистические данные показывают, что такое заболевание встречается у одного новорожденного из семисот малышей. Данный диагноз, как правило, устанавливается специалистом еще в родильном доме, на сроке 3-5 дней жизни новорожденного ребенка.
Для того, чтобы подтвердить данный диагноз, проводится такая процедура, как исследование кариотипа. Она заключается в исследовании набора хромосом у новорожденного ребенка. Ребенок, который болеет , имеет семь хромосом, что на одну больше, чем у здорового человека. Такое заболевание встречается, как у мальчиков, так и у девочек, пол в данном случае не играет никакой роли.
Болезнь Шершевского-Тернера
Данное заболевание характерно только для детей женского пола. Первые признаки данной генетической патологии можно обнаружить в возрасте 10-12 лет.
Как правило, на затылке волосы растут очень медленно, причем, они имеют глубоко – посаженный корень. В возрасте 15 – 16 лет, а то и старше, у девочек отсутвуют , именно это и является причиной обращения к специалисту. С возрастом, заболевание может стать причиной проявления некоторых проблем с умственным развитием ребенка. Генетическая структура болезни Шершевского-Тернера у девочек характеризуется отсутствием одной Х хромосомы.
Болезнь Клайнфельтера
Болезнь Клайнфельтера — это генетическая патология, которая проявляется только у мальчиков. Первые признаки заболевания можно будет обнаружить при достижении ребенком 15 – 16 – летнего возраста.
Первые признаки:

При исследовании на хромосомы болезнь Клайнфельтера характеризуется их увеличенным количеством: одной хромосомой Х больше. В некоторых случаях, могут дополнительно присутствовать и другие хромосомы: У, ХХ, ХУ.
Многофакторные генетические заболевания
Многофакторные генетические заболевания представляют собой генетические патологии, которые могут проявиться у новорожденного ребенка в любой семье.
В данном случае причиной развития таких заболеваний выступают не только генетические отклонения, но и ряд посторонних факторов, например, плохая экология, нарушенный ритм жизни родителей.
К числу таких заболеваний относится: ишемическая болезнь сердца, заболевания желудка, а также проблемы с кровеносной системой.
Врожденные дефекты, которые относятся к многофакторным генетическим заболеваниям — это заячья губа, волчья пасть и расщепление позвоночника.
В настоящее время все многие патологии можно выявить с помощью современного оборудования: ультразвуковое исследование плода сможет показать многие аномалии в развитии ребенка.
Генетические заболевания – это редкие и сложные по своей природе болезни, которые практически не поддаются лечению из-за нарушений на геном уровне. Поэтому специалисты рекомендуют при планировании ребенка заранее проходить консультацию с генетиком во избежание проблем в будущем.
О генетических болезнях расскажут специалисты на видео:
Заметили ошибку? Выделите ее и нажмите Ctrl+Enter , чтобы сообщить нам.
Понравилось? Лайкни и сохрани у себя на страничке!
Рождение ребенка - самое счастливое событие для каждой семейной пары. Ожидание встречи с малышом часто бывает омрачено тревожными мыслями по поводу его здоровья и правильного развития. В большинстве случаев тревоги молодых родителей оказываются напрасными, но порой судьба обходится с еще не рожденным крохой довольно сурово: малыш получает от мамы и папы не только цвет волос, разрез глаз и милую улыбку, но и различные наследственные болезни.
По данным медицинской статистики, вероятность рождения ребенка с наследственной патологией для каждой будущей мамы составляет 3–5%. Например, вероятность рождения детей с синдромом Дауна составляет 1:700. Наиболее тяжело диагностируются и поддаются дальнейшему лечению редкие, так называемые орфанные, заболевания: несовершенный остеогенез, буллезный эпидермолиз, синдром Менкеса, прогерия и многие другие. Как правило, эти генные наследственные болезни создают угрозу для жизни ребенка, значительно снижают ее продолжительность и качество, приводят к наступлению инвалидности. В нашей стране «редкими» принято считать заболевания, проявляющиеся с частотой 1:10000.
Причины наследственных болезней
 Каждая клетка человеческого организма несет в себе определенный код, заключенный в хромосомах. Всего у человека их 46: из них 22 пары аутосомные, а 23-я пара хромосом отвечает за пол человека. Хромосомы в свою очередь состоят из множества генов, несущих в себе информацию об определенном свойстве организма. Самая первая клетка, образующаяся при зачатии, содержит 23 материнские хромосомы и такое же количество отцовские. Дефект гена или хромосомы и приводит к возникновению генетических нарушений.
Каждая клетка человеческого организма несет в себе определенный код, заключенный в хромосомах. Всего у человека их 46: из них 22 пары аутосомные, а 23-я пара хромосом отвечает за пол человека. Хромосомы в свою очередь состоят из множества генов, несущих в себе информацию об определенном свойстве организма. Самая первая клетка, образующаяся при зачатии, содержит 23 материнские хромосомы и такое же количество отцовские. Дефект гена или хромосомы и приводит к возникновению генетических нарушений.
Существуют различные типы генетических нарушений: дефект одного гена, дефект хромосом и комплексный дефект.
Дефект одного гена может передаваться от одного или обоих родителей. Причем, являясь носителем рецессивного гена, мама и папа могут даже не знать о своем заболевании. К таким заболеваниям относятся прогерия, синдром Менкеса, буллезный эпидермолиз, несовершенный остеогенез. Дефект, передающийся с хромосомой из 23 половой пары, называется Х-сцепленным. Каждый человек наследует от матери Х-хромосому, а вот от отца он может получить Y-хромосому (в этом случае на свет рождается мальчик) или Х-хромосому (появляется девочка). Если на Х-хромосоме мальчика обнаруживается дефектный ген, он не может быть уравновешен второй здоровой Х-хромосомой, а потому появляется вероятность развития патологии. Этот дефект может передаваться от матери-носителя заболевания или формироваться совершенно непредсказуемо.
Дефект хромосом - изменение их структуры и числа. В основном такие дефекты образуются при формировании яйцеклеток и сперматозоидов родителей, хромосомный дефект возникает у зародыша при слиянии этих клеток. Такая патология, как правило, проявляется в виде серьезных нарушений в физическом и умственном развитии.
Комплексные дефекты возникают в результате воздействия на ген или группу генов факторов внешней среды. Механизм передачи данных заболеваний все еще не изучен до конца. По предположениям медиков ребенок наследует от родителя особую чувствительность к определенным факторам окружающей среды, под влиянием которых в итоге может развиться заболевание.
Диагностика в до родовый период
 Наследственные болезни детей могут быть выявлены еще в дородовом периоде. Так, в последнее время во многих консультациях тест, определяющий уровень содержания гормонов АФП, эстрогена и ЧХГ, проводится всем женщинам между и 18 неделями беременности . Он помогает определить патологии развития ребенка вследствие хромосомных дефектов. Стоит отметить, что данный скрининг позволяет выявить лишь часть генетических нарушений, в то время как современная классификация наследственных болезней представляет собой сложную систему, включающую около двух тысяч болезней, состояний и синдромов.
Наследственные болезни детей могут быть выявлены еще в дородовом периоде. Так, в последнее время во многих консультациях тест, определяющий уровень содержания гормонов АФП, эстрогена и ЧХГ, проводится всем женщинам между и 18 неделями беременности . Он помогает определить патологии развития ребенка вследствие хромосомных дефектов. Стоит отметить, что данный скрининг позволяет выявить лишь часть генетических нарушений, в то время как современная классификация наследственных болезней представляет собой сложную систему, включающую около двух тысяч болезней, состояний и синдромов.
Будущим родителям следует иметь ввиду, что на основании результатов данного анализа не диагностируется определенное заболевание, а лишь определяется его вероятность и принимается решение о необходимости дополнительных обследований.
Амниоцентез - процедура, во время которой врач с помощью тонкой и длинной иглы делает забор амниотической жидкости, проникая в матку женщины через брюшную стенку. Предварительно женщина направляется на ультразвуковое исследование для определения положения плода и наилучшего места введения иглы. Иногда УЗИ проводится прямо во время процедуры амниоцентеза.
Это исследование позволяет выявить множество хромосомных дефектов, определить степень развития легких ребенка (в случае необходимости родов до запланированного срока), точно определить пол ребенка (при угрозе возникновения заболеваний, связанных с определенным полом). Исследование полученной жидкости занимает несколько недель. Недостаток этой процедуры заключается в том, что она может проводиться на сроке беременности свыше 16 недель , а значит, времени для принятия решения о прерывании беременности у женщины остается очень мало. К тому же в отличие от первого триместра, аборт на таком большом сроке -крайне опасная процедура как для физического, так и для психического здоровья женщины. Риск самопроизвольного аборта после данного исследования колеблется от 0,5 до 1%.
С помощью исследования хориона (ткани, окружающей плод на раннем сроке беременности) также можно определить генетические нарушения у плода, в том числе и диагностировать довольно редкие болезни, такие как буллезный эпидермолиз, несовершенный остеогенез. Во время этой процедуры врач через влагалище вводит тонкую трубку в матку женщины. Кусочки ворсинок хориона всасываются через трубку, а затем отправляются на анализ. Данная процедура безболезненна и может проводиться уже на 9 неделе беременности, результаты исследования будут готовы через один-два дня. Несмотря на очевидные преимущества, данная процедура не слишком востребована из-за высокого риска самопроизвольных абортов (2–3%) и различных нарушений течения беременности.
Показаниями для исследования хориона и амниоцентеза являются:
- возраст будущей мамы больше 35 лет;
- хромосомные дефекты у одного или обоих родителей;
- рождение у семейной пары ребенка с хромосомными дефектами;
- будущие мамы, в семьях которых были Х-сцепленные заболевания.
Если проведенные исследования подтвердили наличие генетического нарушения, родителям, взвесив все «За» и «Против», предстоит сделать, пожалуй, самый трудный выбор в их жизни: сохранить или прервать беременность, поскольку лечение наследственных болезней на данномэтапе, к сожалению, невозможно.
Диагностика после рождения ребенка
 Диагностировать редкие генные наследственные болезни можно на основании лабораторных исследований. Вот уже несколько лет во всех родильных домах на пятый день после рождения малыша проводится скрининг новорожденных, в ходе которого диагностируется ряд редких наследственных заболеваний: фенилкетонурия, гипотиреоз, муковисцидоз, галактоземия и адрено-генитальный синдром.
Диагностировать редкие генные наследственные болезни можно на основании лабораторных исследований. Вот уже несколько лет во всех родильных домах на пятый день после рождения малыша проводится скрининг новорожденных, в ходе которого диагностируется ряд редких наследственных заболеваний: фенилкетонурия, гипотиреоз, муковисцидоз, галактоземия и адрено-генитальный синдром.
Остальные заболевания диагностируются на основании симптомов и признаков, которые могут проявляться как в период новорожденности, так и спустя много лет после рождения. Симптомы буллезного эпидермолиза и несовершенногоостеогенезав большинстве случаев проявляются сразу после рождения, а диагноз прогерия чаще всего ставится только на 2–3 году жизни ребенка.
Рядовому педиатру очень сложно бывает распознать редкие болезни, врач может просто не заметить их симптомы во время обычного приема. Именно поэтому маме нужно быть очень внимательной по отношению к собственному ребенку и обращать внимание на угрожающие признаки: не по возрасту моторика, появление судорог, недостаточный набор веса, неестественный цвет и запах испражнений. Также поводом для тревоги должно стать резкое увеличение или замедление процесса роста ребенка, это может свидетельствовать о наличии такого заболевания, как карликовость. При появлении подобных симптомов родителям необходимо обязательно обратиться к врачу, настаивая на тщательном обследовании ребенка, ведь своевременная диагностика наследственных болезней и подбор правильной программы лечения могут помочь сохранить здоровье, а порой и жизнь малыша.
Как лечат генетические заболевания?
Хотя большинство наследственных болезней не поддается лечению, современная медицина в состоянии значительно увеличить продолжительность жизни больных детишек, а также улучшить ее качество. На сегодняшний день такие заболевания - не приговор, а скорее образ жизни, позволяющей ребенку нормально развиваться при условии получения необходимого лечения: прием лекарственных препаратов, гимнастика, специальные диеты. Причем чем раньше удается диагностировать, тем более успешно проходит лечение наследственных болезней.
В последнее время все чаще применяются методы пренатального (дородового) лечения: с помощью лекарственных препаратов и даже хирургических операций.
Болезнь ребенка - тяжелое испытание для всей семьи. В этих условиях родителям очень важна поддержка родственников и общение с другими мамами и папами, оказавшимися в аналогичной ситуации. Большую помощь таким семьям оказывают различные сообщества родителей с детишками, имеющими редкие генетические заболевания.
Как предотвратить наследственные заболевания?
Грамотное планирование беременности, основным направлением которого является профилактика наследственных болезней, поможет избежать рождения больного ребенка. Родителям, входящим в группу риска, стоит обязательно посетить врача-генетика:
- возраст родителей −35 лет и выше;
- наличие одного или более детей с наследственным заболеванием;
- редкие заболеванияу супругов или их близких родственников;
- пары, беспокоящиеся по поводу рождения здорового ребенка.
Консультант-генетик на основании данных медицинского обследования, а также информации об истории семьи, заболеваниях, которыми болели родственники, наличии абортов и выкидышей, рассчитывает вероятность рождения ребенка с генетическим заболеванием. Случается, что пара, имеющая большие шансы родить больного ребенка, отказывается от этих планов в данном союзе, а с другими партнерами обзаводятся совершенно здоровыми детишками.
Девочки! Давайте делать репосты.
Благодаря этому к нам заглядывают специалисты и дают ответы на наши вопросы!
А еще, вы можете задать свой вопрос ниже. Такие как вы или специалисты дадут ответ.
Спасибки;-)
Всем здоровых малышей!
Пс. Мальчиков это тоже касается! Просто девочек тут больше;-)
Понравился материал? Поддержите - сделайте репост! Мы стараемся для вас;-)