Това се пресича! Оригиналът е взет от eka_tyryshkina Грешките на природата: хора с редки болести.
Тук се разболях онзи ден, както винаги, тъй като отивахме само там, където трябваше да посетим, болен съм! Толи, болестта ми отговаря на планираността на събитието, а също и на нещо, но е добре, че не реагира на работата. Като цяло болестта не е лесна за мен)
И ето, че съм болен в късен час, след като вече е свършил цялата работа, препрочитам и разглеждам всички интересни уебсайтове, изведнъж не очаквах да разбера за най-редките язви на планетата и знаете толкова много интересно и шокиращо !!!
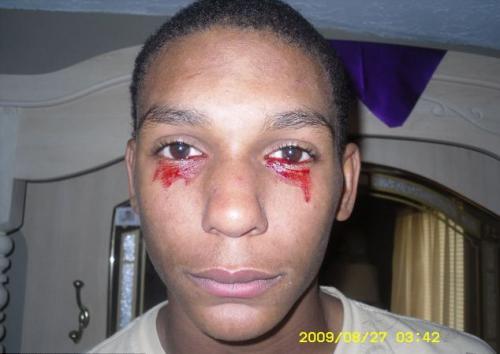
haemolacria
("Кървави сълзи") се наблюдава в един човек на милион.Кръвта, вместо сълза, изведнъж започва да тече от очите и това може да продължи около час. През деня пациентът излива кървави сълзи от 3 до 20 пъти.
Точната причина за това заболяване не е напълно изяснена и следователно не може да бъде лекувана. Медицинските специалисти досега са представили версията, че хемолакрията е едно от кръвните заболявания или тумори.
На снимката - 15-годишен Калвино Инман (Тенеси, САЩ)

Вампирен синдром
Диагностициран с "Вампирски синдром"
(ектодермална дисплазия) в света има само 7 хиляди души.
В допълнение към смъртоносната бледа кожа и остри кучешки зъби (при липса на част от зъбите), пациентите имат рядка и тънка коса, способността им да се потят се намалява, следователно тялото им е предразположено към прегряване. Симптомите се проявяват в детска възраст, но е възможно да се идентифицира заболяването още на етапа на бременността с помощта на генетични тестове.
Момчетата са принудени да носят слънчеви очила и да използват слънцезащитни продукти, когато излизат навън, тъй като не могат да бъдат под пряка слънчева светлина.В същото време физическото развитие и физическата активност остават нормални.Самата болест е нелечима, само симптомите могат да бъдат коригирани. По-специално, можете да възстановите нормалната форма на зъбите.
Болестта на Саймън е диагностицирана в ранна детска възраст. Когато Манди била бременна за втори път, тя била предупредена, че второто дете може да има същото заболяване.Въпреки това, Саймън се разви и се разви добре, така че родителите поеха този риск.
Момчетата казват: "Някои деца се смеят на нашия външен вид, но нашите приятели мислят, че е готино."
На снимката - Саймън (13-годишен) и Джордж (11-годишен) Кълън (Съфолк, Великобритания).

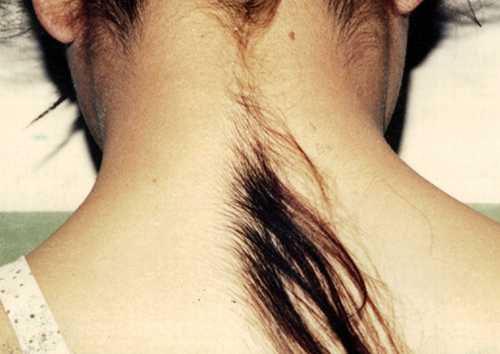
хипертрихоза
("Синдром на Върколака") е заболяване, което се проявява при прекомерен растеж на косата, което не е характерно за тази област на кожата, което не съответства на пола и възрастта. грозота ... Те подават заявления в Книгата на рекордите на Гинес - за да станат известни и да печелят пари ... Китайският Ю Дженхуанг успя да направи това за всички - благодарение на супер-космат, той основава най-популярната рок група в страната си и става милионер.
Не е известно защо се случва тази мутация. И никой все още не е разработил лечение за хипертрихоза. Козметиците могат да премахват косата само за достатъчно дълъг период от време ...
На снимката - 6-годишен Нат Сасуфан (Тайланд), 2007 г.

На снимката - 33-годишният Ю Дженхуанг (Китай), най-хубавият човек на света

Болест на слона
("Протеев синдром", elephantiasis, elephantiasis, elephantiasis) - увеличаване на размера на която и да е част на тялото, дължащо се на болезненото развитие на кожата и подкожната тъкан.
А най-известният пациент е „слонът“ - Джозеф Меррик. За известния британец през 1980 г. режисьорът Дейвид Линч дори прави филм, който е номиниран за Оскар в осем номинации ... Филмът е за човешкото достойнство ... Грим Джон Хърт, който е играл Мерик, е създаден на базата на Лондонската болница. на алкохолизираното тяло на Джозеф Мерик. Неговата подложка всеки ден взимаше актьора 12 часа на ден ...
На снимката - 35-годишен Mendi sellars (United Kingdom)

Аномалия на гена се състои в ускоряване стареене на тялото ,
- progeria - подразделени на детска стая (синдром на Гетчинсън) и възрастен (синдром на Вернер). За първи път за синдром на преждевременно стареене започнаха да говорят преди 100 години. И не е изненадващо, че такива случаи се срещат веднъж в 4-8 милиона бебета. Progeria (от гръцката Pro - преди, gerontos - Elder) е изключително рядко генетично заболяване, което ускорява процеса на стареене около 8-10 пъти. Казано по-просто, едно дете в една година расте с 10-15 години. Осемгодишният мъж изглежда 80 години - със суха набръчкана кожа, оплешивяваща глава ... Тези деца обикновено умират след 13-14 години след няколко сърдечни пристъпи и инсулти на фона на прогресивна атеросклероза, катаракта, глаукома, пълна загуба на зъби и др. И само няколко живеят до 20 години или повече.
Сега само 42 случая на прогерия хора са известни в света ... От тях, 14 души живеят в Съединените щати, 5 в Русия, останалите в Европа ...
В момента съществуват няколко организации, които предоставят помощ на малките стари хора и техните семейства. В интернет има сайтове, посветени на този конкретен проблем, някои от тях са отворени за лекари или социални работници, а други за семействата на пациентите.
На снимката - 24-годишният Леон Бот



38-годишен хората Tree
Деде Косвара, който живее на остров Ява, в Индонезия, стана известен по целия свят заради човешкия папиломен вирус, който обикновено води до появата на малки брадавици, но в случая с индонезийците той е деформирал крайниците му до непознаване.
Проблемът на Деде е, че той има рядко генетично заболяване, което не позволява на имунната му система да задържи растежа на тези брадавици. Следователно, вирусът е бил в състояние да "завладее клетъчния механизъм на нейните кожни клетки", като им дава заповеди да произвеждат голямо количество роговица, от която са били съставени. Dede също имаше нисък брой на белите кръвни клетки.


Заболяване с пеперуди
Хиперпластичната булева епидермолиза е генетично заболяване, което се проявява в първите дни от живота. Всъщност кожата на новороденото е толкова деликатна, че всяко докосване води до появата на рани и мехури. Най-засегнатите области са: лакти, колене, крака, ръце. Получаващата се язва, от която кожата се разтваря на слоеве, не лекува дълго време, освобождава течност от нея. След образуването на голям пурпурен белег.
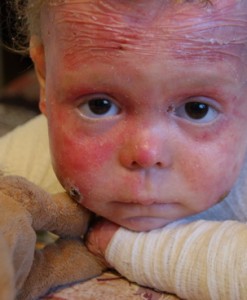
Няма лечение за това заболяване, възможно е само облекчаване на симптомите. Неотдавна историята на Лиза Кунигел, която живее с булева епидермолиза в продължение на почти десет години, разтърси цяла Русия. Няколко пъти на ден, тя се нуждае от превръзки и лечение с антимикробни мехлеми и гелове. В допълнение, всички 9 години Lisa съпътства болката.
Синдром на русалка
Една от най-редките аномалии в развитието е сиреномелия, наречена "синдром на русалка". При този дефект новородените се раждат с подредени крака, които приличат на рибена опашка. Те имат само един бъбрек, няма гениталии. Поради голямото увреждане на вътрешните органи, такива бебета обикновено умират скоро. Заболяването се среща при един от 100 000 новородени. През всичките години на наблюдение, само три бебета са успели да оцелеят. Един от тях беше Шило Пепин.
Шило е роден през 1999 г. и става най-известното дете със синдрома на "русалка". По време на десетте години, в които можеше да живее, тя имаше хиляди приятели по целия свят, които подкрепяха момичето и майка й. Shilo се опита да води пълен живот - тя, като всички обикновени деца, ходи на училище, посещаваше уроци по танци, ходи в увеселителни паркове. Момичето стана известно след участието си в шоуто на Опра Уинфри. Научавайки Chanel направи няколко филма за нея, стотици сайтове в интернет са посветени на нея.

Историята на Сило е удивителна история за чудо. Дете, което през цялото си детство се бореше да оцелее. Малко момиче, което можеше да се наслаждава на всеки ден, въпреки неизлечимата болест.
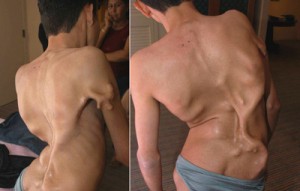
Болестта на Мунхаймер
Фибродиплазията е изключително рядко заболяване. Официалната статистика е следната: 1 пациент на 2,000,000 души. Болестта на Мунхаймер възниква в резултат на генна мутация и при раждането се проявява във външни дефекти. Бебето има усукани големи пръсти, гръбначен стълб. Патологията води до увреждане, ранна смъртност. Когато трябва да се проведат противовъзпалителни процеси, растежът на костите започва да се формира, поради което заболяването често се нарича "второ скелетно заболяване".
Всяко, дори и леко нараняване, може да доведе до развитие на остъкляване в засегнатата област.В момента няма официално лечение на смъртоносната болест. Учените са разработили лекарство, което теоретично може да се бори с болестта. Необходимите клинични проучвания обаче все още не са проведени. Уви, много е трудно да ги държите - в целия свят има не повече от 600 души с болестта на Мюнчамер.
феномен "Линии Блашко" характеризира с наличието на странни ленти в цялото тяло. Линията Блашко е невидим модел, заложен в ДНК. И проявлението на болестта става появата на този модел.
Обикновено моделът на гърба е V-образен, а на гърдите, корема и страните - S-образен.
Причината за заболяването може да бъде мозаицизъм. Във всеки случай, появата на линиите на Блашко по никакъв начин не е свързана с човешката нервна, мускулна и лимфна системи.
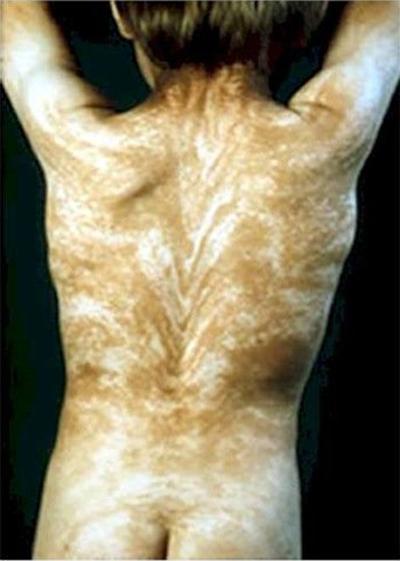
Друга анормална болест е акантозис нигрикансили синдром на синята кожа. Хората с тази диагноза могат да имат синя, индигова, слива или почти лилава кожа.В 60-те години на миналия век в Кентъки живее цялото семейство от „сини” хора. Бяха известни като Сините Фуге. Тази функция е предавана от поколение на поколение.


Около 6% от жителите на света страдат от редки болести и този брой продължава да нараства. Всички уникални болести имат различно естество, но по-голямата част от феноменалните заболявания са свързани с генетични аномалии и инфекции.
Генетичните заболявания са уникални, тъй като не зависят от начина на живот на човека, не могат да бъдат осигурени от просто преставане да ядат мазни храни или да започват да тренират сутрин. Те произтичат от мутации и могат да бъдат предавани от поколение на поколение.
Рядко наследствено заболяване, при което човек умира от неспособност да спи. Досега тя се чества само в 40 семейства по целия свят. Смъртоносно безсъние обикновено настъпва между 30 и 60 години (най-често след 50 години) и продължава от 7 до 36 месеца. С напредването на заболяването пациентът страда от по-тежки нарушения на съня и никакви хапчета за сън не му помагат. На първия етап безсънието е съпроводено с пристъпи на паника и фобии, при втория етап се добавят халюцинации и повишено изпотяване. В третия етап на заболяването, човекът напълно губи способността си да спи и започва да изглежда много по-възрастен от годините си. След това се развива деменция и пациентът умира - обикновено от изтощение или пневмония.

Нарколепсийно-катаплексичният синдром, който се характеризира с внезапни пристъпи на сън и релаксация на мускулите на тялото, също има генетична природа и се причинява от нарушения на фазата на бързото сън. Това се случва много по-често от фаталното семейно безсъние: 40 на всеки 100 хиляди души, еднакво при мъжете и жените. Човек, страдащ от нарколепсия, може внезапно да заспи за няколко минути в средата на деня. “Спящите атаки” напомнят за REM съня и могат да се случват много често: до 100 пъти на ден, с или без главоболие, което ги предшества. Често те се провокират от бездействие, но могат да се появят в напълно неподходящо време: по време на полов акт, спортуване, шофиране. Човек се събужда отпочинал.
![]()
Синдромът на Yuner Tan (SYT) се характеризира предимно с факта, че хората, страдащи от него, ходят на четири крака. Той е открит от турски биолог Юнер Тан, след като е изучил пет члена на семейството на Улас в селска Турция. Най-често хората с ФТК използват примитивна реч и имат вродена церебрална недостатъчност. През 2006 г. за семейството на Ulas е заснет документален филм, озаглавен „Семейството, ходещо на четири крака“. Тан го описва по следния начин: „Генетичната природа на синдрома предполага обратен етап в еволюцията на човека, най-вероятно причинен от генетична мутация, обратната на прехода от квадропедализъм (ходене на четири крайника) към двуполярност (ходене по две). В този случай, синдромът съответства на теорията на периодичното равновесие.

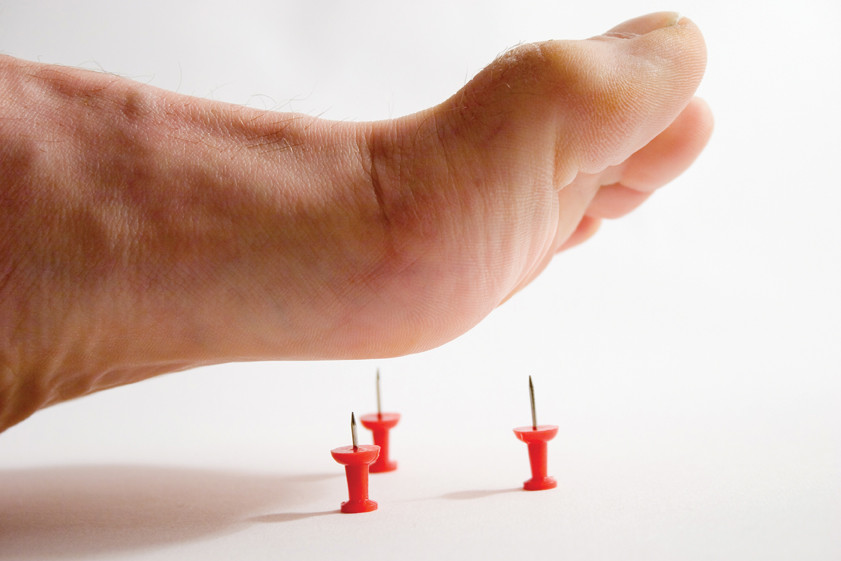
Една от най-редките заболявания в света: този тип невропатия се диагностицира при двама от един милион души. Аномалията е причинена от увреждане на периферната нервна система, което се дължи на прекомерното количество на гена PMP22. Основният признак за развитие на наследствената сензорна невропатия от първия тип е загубата на чувствителност на ръцете и краката. Човек престава да изпитва болка и чувства промяна в температурата, което може да доведе до некроза на тъканите, например, ако не разпознаете фрактура или друга вреда навреме. Болката е една от реакциите на тялото, която сигнализира за "неизправности", така че загубата на чувствителност към болка е изпълнена с твърде късно откриване на опасни заболявания, било то инфекции или язви.
Хората, страдащи от това необичайно заболяване, изглеждат много по-възрастни от възрастта си, поради което понякога се наричат \u200b\u200b"обратния синдром на Бенджамин Бътън". Поради наследствена генетична мутация, а понякога и в резултат на употребата на определени лекарства в организма, се нарушават автоимунните механизми, което води до бърза загуба на подкожни мастни резерви. Мастната тъкан на лицето, шията, горните крайници и тялото най-често страда, което води до бръчки и гънки. Досега са потвърдени само 200 случая на прогресивна липодистрофия и се развиват главно при жени. При лечението лекарите използват инсулин, „стягане” на лицето и инжекции с колаген, но това дава само временен ефект.

Хипертрихозата се нарича също „синдром на върколака” или „синдром на Абрамс”. Тя се проявява само в един човек от един милиард и само 50 случая от средновековието са документирани. Хората, страдащи от хипертрихоза, се характеризират с прекомерно количество коса по лицето, ушите и раменете. Това се дължи на разпадането на връзките между епидермиса и дермата по време на образуването на космените фоликули при тримесечен плод. Като правило, сигналите от получената дерма "информират" фоликулите за тяхната форма. На свой ред фоликулите също сигнализират на кожните слоеве, че вече има един фоликул в тази област и това води до факта, че космите по тялото растат приблизително на същото разстояние един от друг. В случай на хипертрихоза, тези връзки са счупени, което води до образуване на твърде гъста коса на тези части на тялото, където не трябва да бъде.

Ако някога сте чували за припадък от коза, тогава знаете как изглежда вродената миотония - поради мускулни спазми, човек замръзва за известно време. Причината за вродена (вродена) миотония е генетично заболяване: поради мутация функционирането на хлорните канали на скелетните мускули е нарушено. Мускулната тъкан е "объркана", произтичат контракции и релаксация, а патологията може да засегне мускулите на краката, ръцете, челюстите и диафрагмата.

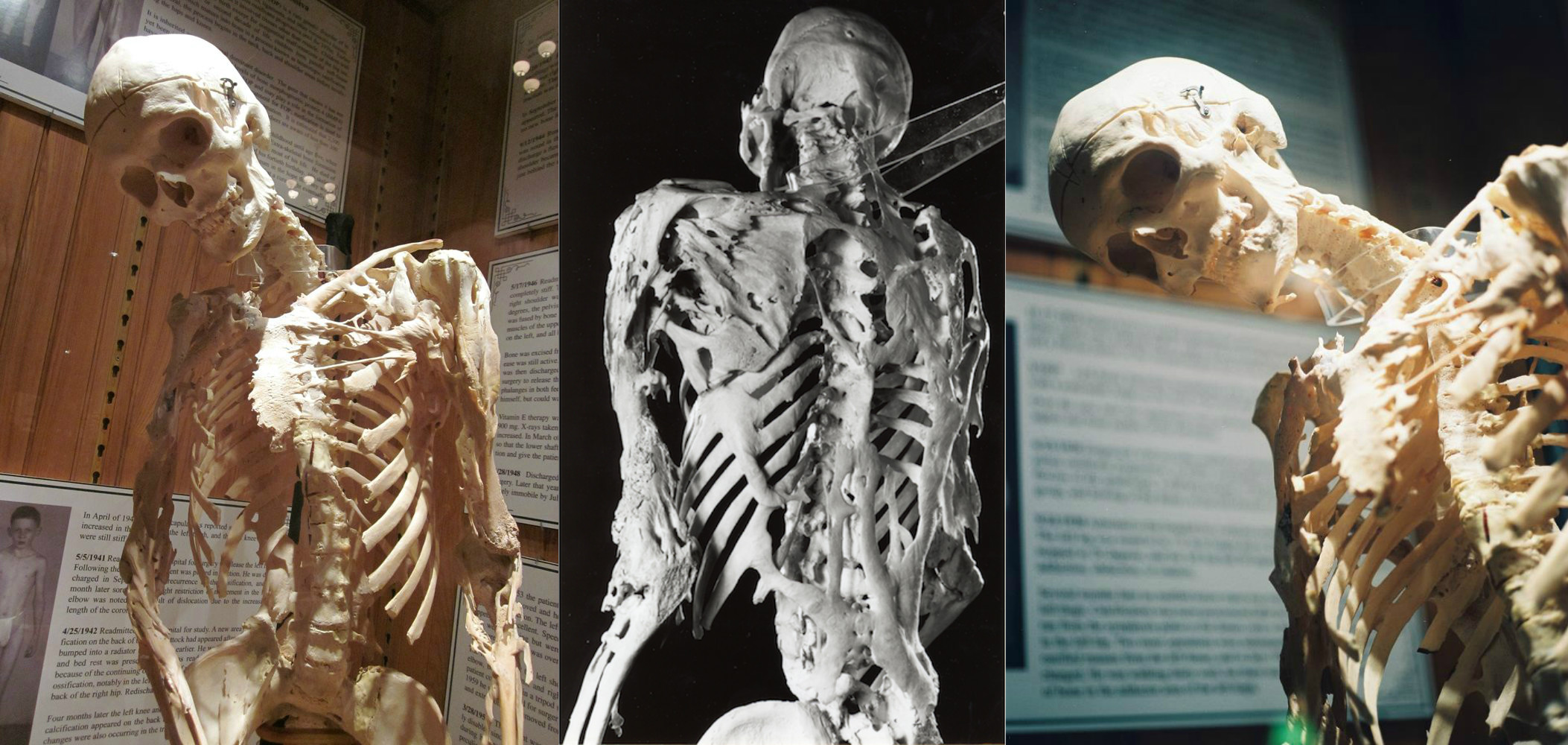
Рядко генетично заболяване, при което тялото започва да образува нови кости - осифицирани - в погрешни места: вътре в мускулите, сухожилията, сухожилията и други съединителни тъкани. Всяко нараняване може да доведе до образуването им: контузия, нарязване, фрактура, интрамускулна инжекция или хирургична намеса. Поради това е невъзможно да се премахнат осефиатите: след операцията костта може да стане по-силна. Физиологично, осификатите не се различават от обикновените кости и могат да издържат на значителни натоварвания, само че не са на правилното място.
FOP се причинява от мутация в ACVR1 / ALK2 гена, който кодира костен морфогенетичен протеинов рецептор. Тя се предава на човек по наследство от един от родителите, ако той също е болен. Да бъдеш носител на това заболяване е невъзможно: пациентът или е болен, или не. Досега FOP е сред нелечимите болести, но в момента се провежда втора поредица от изпитвания на лекарство, наречено паловаротен, което ви позволява да блокирате гена, отговорен за патологията.
Това наследствено заболяване на кожата се проявява в повишената чувствителност на човек към ултравиолетовите лъчи. Това се случва поради мутация на протеини, отговорни за възстановяване на увреждане на ДНК, което се проявява при излагане на ултравиолетова радиация. Първите симптоми обикновено се появяват в ранна детска възраст (до 3 години): когато детето е на слънце, той има сериозни изгаряния след няколко минути излагане на слънце. Също така, болестта се характеризира с появата на лунички, суха кожа и неравномерна промяна в цвета на кожата. Според статистиката, хората с пигментна ксеродерма са изложени на по-голям риск от развитие на рак: при липса на подходящи превантивни мерки около половината от децата, страдащи от ксеродерма, развиват определени видове рак на десетгодишна възраст. Има осем вида заболявания с различна тежест и симптоми. Според европейски и американски лекари болестта се среща в около четири милиона души.

Любопитно име за болестта, нали? Въпреки това, има един научен термин за този "болки" - десквамационен глосит. Географският език се проявява при около 2,58% от хората и най-често заболяването има хронични свойства и се влошава след хранене, по време на стрес или хормонален стрес. Симптомите се проявяват във външния вид на езика на избелени гладки петна, наподобяващи острови, тъй като болестта има такъв необичаен псевдоним, и с течение на времето някои от “островите” променят формата и местоположението си, в зависимост от това кои от вкусовите пъпки, разположени на езика, лекуват, и, напротив, са раздразнени.
Географският език на практика е безвреден, ако не вземете под внимание свръхчувствителност към пикантна храна или някакъв дискомфорт, който може да причини. Причините за това заболяване не са известни на медицината, но има доказателства за генетична предразположеност към неговото развитие.
Не само външни черти и черти на характера могат да бъдат прехвърлени на детето от неговите биологични родители, но и редица здравословни проблеми.
Наследствените заболявания са редки, но по правило това са доста сериозни заболявания, които на практика не подлежат на лечение.
Всеки ген на човешкото тяло съдържа уникална ДНК, има свой собствен уникален код на специфична черта.
В тази ситуация е необходимо да се потърси помощ от лекар - генетика, да се подложи на генетично консултиране, да се установи степента на риск от конкретно генетично заболяване.
Болест на Даун
Днес, едно от най-често срещаните заболявания

е наследствена, е болест на Даун. Статистиката показва, че това заболяване се среща при едно новородено от седемстотин бебета. Тази диагноза обикновено се установява от специалист, който все още се намира в родилния дом, за период от 3-5 дни от живота на новородено дете.
За да се потвърди тази диагноза, се провежда процедура като кариотипно изследване. Това е изследване на множеството хромозоми при новородено бебе. Детето, което е болно, има седем хромозоми, един повече от здрав човек. Това заболяване възниква както при момчета, така и при момичета, като в този случай сексът не играе никаква роля.
Болест на Шершевски-Търнър
Това заболяване е характерно само за женските деца. Първите признаци на тази генетична патология могат да бъдат открити на възраст 10-12 години.
Като правило, на гърба на главата косата расте много бавно и освен това те имат дълбоко вкоренен корен. На възраст от 15 до 16 години и дори по-възрастни, момичетата не съществуват и именно това е причината за търсене на специалист. С възрастта болестта може да предизвика проявление на някои проблеми с умственото развитие на детето. Генетичната структура на болестта на Шершевски-Търнър при момичетата се характеризира с липсата на една Х-хромозома.
Болест на Klinefelter
Болестта на Klinefelter е генетично заболяване, което се проявява само при момчета. Първите признаци на заболяването могат да бъдат открити, когато детето навърши 15-16 години.
Първите признаци са:

При изследване на хромозомите болестта на Klinefelter се характеризира с увеличен брой: още една Х хромозома. В някои случаи могат да присъстват и други хромозоми: Y, XX, XY.
Мултифакторни генетични заболявания
Мултифакторните генетични заболявания са генетични патологии, които могат да възникнат при новородено дете във всяко семейство.
В този случай причината за развитието на такива заболявания е не само генетичните аномалии, но и редица външни фактори, например лоша екология, нарушен ритъм на живота на родителите.
Тези заболявания включват: коронарна болест на сърцето, заболявания на стомаха, както и проблеми с кръвоносната система.
Вродени дефекти, които са свързани с мултифакторни генетични заболявания, са цепната устна, цепнато небце и спина бифида.
В момента всички многобройни патологии могат да бъдат идентифицирани с помощта на съвременна апаратура: ултразвуково изследване на плода ще може да разкрие много аномалии в развитието на детето.
Генетичните заболявания са редки и комплексни по характер, заболявания, които почти не се лекуват поради нарушения на геномното ниво. Ето защо експертите препоръчват при планиране на дете предварително да преминат консултация с генетик, за да се избегнат проблеми в бъдеще.
Експерти ще разкажат за генетичните заболявания на видео:
Забелязахте грешка? Изберете го и щракнете върху Ctrl + Enterда ни уведомите.
Харесва ли ви? Laykni и запишете на вашата страница!
радостно събитие - най-щастливото събитие за всяка двойка. Чакането на среща с бебето често се замъглява от смущаващи мисли за неговото здраве и правилно развитие. В повечето случаи тревогите на младите родители са напразни, но понякога съдбата се отнася много сериозно към нероденото бебе: бебето получава от мама и татко не само цвят на косата, форма на очите и сладка усмивка, но и различни наследствени заболявания.
Според медицинската статистика вероятността да има дете с наследствена патология за всяка бременна майка е 3-5%. Например, вероятността да имаш деца със синдром на Даун е 1: 700. Редки, т.нар. Болести с редки заболявания, като остеогенеза imperfecta, булезна епидермолиза, синдром на Menkes, прогерия и много други, са най-тежко диагностицирани и могат да бъдат допълнително лекувани. Като правило, тези генетични наследствени заболявания представляват заплаха за живота на детето, значително намаляват неговата продължителност и качество и водят до появата на увреждания. В нашата страна "рядко" се считат болести, които се проявяват с честота 1: 10,000.
Причини за наследствени заболявания
 Всяка клетка на човешкото тяло носи определен код, затворен в хромозомите. Човек има 46 от тях: 22 от тях са автозомни, а 23-та двойка хромозоми отговаря за пола на човека. Хромозомите от своя страна се състоят от много гени, които носят информация за дадено свойство на организма. Първата клетка, образувана по време на зачеването, съдържа 23 майчински хромозоми и същия брой бащини. Дефект на гена или хромозома и води до появата на генетични нарушения.
Всяка клетка на човешкото тяло носи определен код, затворен в хромозомите. Човек има 46 от тях: 22 от тях са автозомни, а 23-та двойка хромозоми отговаря за пола на човека. Хромозомите от своя страна се състоят от много гени, които носят информация за дадено свойство на организма. Първата клетка, образувана по време на зачеването, съдържа 23 майчински хромозоми и същия брой бащини. Дефект на гена или хромозома и води до появата на генетични нарушения.
Съществуват различни видове генетични нарушения: дефект на един ген, дефект на хромозома и сложен дефект.
Дефект на единичен генможе да се предава от един или двамата родители. Освен това, като носител на рецесивния ген, мама и татко може и да не са наясно със своята болест. Такива заболявания включват прогерия, синдром на Menkes, булева епидермолиза, остеогенеза imperfecta. Дефект, предаван с хромозома от 23 полови двойки, се нарича Х-свързан. Всеки човек наследява Х-хромозомата от майката, но от бащата може да получи Y-хромозомата (в този случай се ражда момче) или Х-хромозомата (появява се момиче). Ако на Х-хромозомата на момчето е открит дефектен ген, той не може да бъде балансиран от втората здрава Х-хромозома и следователно се появява вероятността от развитие на патология. Този дефект може да бъде предаден от майката на носителя на болестта или форма напълно непредсказуема.
Хромозомен дефект - да променят структурата и броя им. По принцип такива дефекти се образуват при образуването на яйцата и сперматозоидите на родителите, хромозомният дефект се среща в ембриона по време на сливането на тези клетки. Подобна патология, като правило, се проявява под формата на сериозни нарушения във физическото и психическото развитие.
Сложни дефекти възникват в резултат на излагане на ген или група от гени на фактори на околната среда. Механизмът на предаване на тези заболявания все още не е напълно изяснен. Според предположенията на лекарите, детето наследява от родителя специална чувствителност към определени фактори на околната среда, под влияние на които може да се развие болестта.
Диагноза в преждевременния период
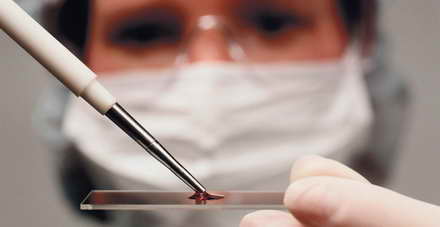 Наследствени заболявания при деца могат да бъдат открити дори в пренаталния период. Така наскоро в много консултации се провежда тест за определяне на нивото на съдържанието на хормоните AFP, естроген и ChCH за всички жени между и 18-седмична бременност , Той помага да се определи патологията на развитието на детето поради хромозомни дефекти. Трябва да се отбележи, че този скрининг позволява да се разкрие само част от генетичните заболявания, докато съвременната класификация на наследствените заболявания е сложна система, включваща около две хиляди заболявания, състояния и синдроми.
Наследствени заболявания при деца могат да бъдат открити дори в пренаталния период. Така наскоро в много консултации се провежда тест за определяне на нивото на съдържанието на хормоните AFP, естроген и ChCH за всички жени между и 18-седмична бременност , Той помага да се определи патологията на развитието на детето поради хромозомни дефекти. Трябва да се отбележи, че този скрининг позволява да се разкрие само част от генетичните заболявания, докато съвременната класификация на наследствените заболявания е сложна система, включваща около две хиляди заболявания, състояния и синдроми.
Бъдещите родители трябва да имат предвид, че въз основа на резултатите от този анализ дадена диагноза не се диагностицира, а само определя нейната вероятност и решава дали са необходими допълнителни изследвания.
амниоцентеза- процедура, при която лекарят с помощта на тънка и дълга игла прави проби от околоплодната течност, прониквайки през матката на жената през коремната стена. Преди това жената е изпратена на ултразвук, за да определи положението на плода и най-доброто място за поставяне на иглата. Понякога ултразвукът се извършва правилно по време на процедурата на амниоцентеза.
Това изследване позволява да се идентифицират много хромозомни дефекти, да се определи степента на развитие на белите дробове на детето (ако е необходимо, доставка преди определеното време), да се определи точно пола на детето (ако съществува риск от заболявания, свързани с определен пол). Изследването, получено от течност, отнема няколко седмици. Недостатъкът на тази процедура е, че може да се извърши по време на продължителността на бременността 16 седмици Това означава, че жената има много малко време да вземе решение за аборт. Освен това, за разлика от първото тримесечие, абортът при такъв дългосрочен план е изключително опасна процедура както за физическото, така и за психичното здраве на жената. Рискът от спонтанен аборт след това проучване варира от 0,5 до 1%.
Използвайки изследването на хориона (тъканите около плода в ранна бременност), също е възможно да се идентифицират генетични аномалии в плода, включително диагностициране на съвсем редки болести като булева епидермолиза, остеогенеза imperfecta. По време на тази процедура лекарят вмъква тънка тръба в матката на жената през вагината. Части от хорионни вълни се всмукват през тръба и след това се изпращат за анализ. Тази процедура е безболезнена и може да се извърши вече Седмица 9 резултатите от бременността ще бъдат на разположение за един до два дни. Въпреки очевидните предимства, тази процедура не е много популярна поради високия риск от спонтанни аборти (2-3%) и различни нарушения на хода на бременността.
Показания за изследване на хорион и амниоцентеза са:
- възрастта на бъдещата майка е над 35 години;
- хромозомни дефекти при един или двамата родители;
- раждането на брачна двойка от дете с хромозомни дефекти;
- бъдещи майки, в чиито семейства има Х-свързани заболявания.
Ако проведените изследвания потвърдят наличието на генетично заболяване, родителите, след претегляне на всички „За” и „Против”, трябва да направят, може би, най-трудния избор в живота си: да спасят или да прекратят бременността, тъй като лечението на наследствени заболявания на този етап, за съжаление, е невъзможно.
Диагностика след раждане
 Диагностицирането на редки наследствени генетични заболявания може да се основава на лабораторни изследвания. В продължение на няколко години във всички родилни домове, на петия ден след раждането на бебето, се извършва скрининг на новородено, по време на което се диагностицират редица редки наследствени заболявания: фенилкетонурия, хипотиреоидизъм, кистозна фиброза, галактоземия и адреногенитален синдром.
Диагностицирането на редки наследствени генетични заболявания може да се основава на лабораторни изследвания. В продължение на няколко години във всички родилни домове, на петия ден след раждането на бебето, се извършва скрининг на новородено, по време на което се диагностицират редица редки наследствени заболявания: фенилкетонурия, хипотиреоидизъм, кистозна фиброза, галактоземия и адреногенитален синдром.
Останалите заболявания се диагностицират въз основа на симптоми и признаци, които могат да се появят както в неонаталния период, така и в много години след раждането. Симптомите на епидермолиза и несъвършена остеогенеза в повечето случаи се появяват непосредствено след раждането, а прогерията най-често се диагностицира само след 2-3 години от живота на детето.
Много е трудно за един обикновен педиатър да разпознава редки болести, лекарят може просто да не забележи техните симптоми по време на нормален прием. Ето защо майката трябва да бъде много внимателна към собственото си дете и да обръща внимание на заплашителните признаци: не поради възрастта на подвижността, появата на гърчове, недостатъчното наддаване на тегло, неестествения цвят и миризмата на фекалии. Също така, рязко увеличаване или забавяне на процеса на растеж на детето трябва да бъде причина за тревога, това може да означава наличието на такава болест като джуджето. Когато тези симптоми се появят, родителите трябва винаги да се консултират с лекар, настоявайки за задълбочено изследване на детето, защото навременната диагностика на наследствени заболявания и изборът на правилната програма за лечение могат да помогнат за запазването на здравето, а понякога и за живота на бебето.
Как се лекуват генетичните заболявания?
Въпреки че повечето наследствени заболявания не реагират на лечението, съвременната медицина е в състояние значително да увеличи продължителността на живота на болните деца, както и да подобри нейното качество. Към днешна дата такива болести не са присъда, а по-скоро начин на живот, който позволява на детето да се развива нормално, при условие че получи необходимото лечение: медикаменти, гимнастика, специални диети. Освен това, колкото по-рано е възможно да се диагностицира, толкова по-успешно е лечението на наследствени заболявания.
Напоследък все повече се използват методите на пренаталното (пренатално) лечение: с помощта на наркотици и дори операции.
Болестта на детето - труден тест за цялото семейство. При тези обстоятелства родителите са много важни да подкрепят роднини и да общуват с други майки и татковци, които се намират в подобна ситуация. Голяма помощ за тези семейства се осигурява от различни общности на родители с деца, които имат редки генетични заболявания.
Как да се предпазим от наследствени заболявания?
Правилното планиране на бременността, чийто основен фокус е превенцията на наследствени заболявания, ще помогне да се избегне раждането на болно дете. Родителите в риск трябва определено да посетят генетик:
- родители на възраст -35 и повече години;
- с едно или повече деца с наследствено заболяване;
- редки болести на съпрузи или близки роднини;
- двойки, които се притесняват да имат здраво бебе.
Генетичен консултант, базиран на данни от медицински преглед, както и информация за фамилна анамнеза, болести, които роднините са претърпели, аборти и спонтанни аборти, изчислява вероятността да има дете с генетично заболяване. Случва се, че двойка с големи шансове да роди болно дете отказва тези планове в този съюз, а с други партньори придобиват напълно здрави деца.
Момичета! Нека направим репост.
Благодарение на това експертите се отзовават и дават отговори на нашите въпроси!
Също така можете да зададете въпроса си по-долу. Такива вие или експерти ще отговорите.
Spasibki ;-)
Всички здрави деца!
Пс. Момчетата също са загрижени! Само момичета тук повече ;-)
Хареса ли ти материалът? Подкрепа - направете репост! Ние се опитваме за вас ;-)