|
DETERMINATION OF THE PRIMARY PROTEIN STRUCTURE |
Determination of the primary structure of the protein will require a preliminary series of operations. The protein must be thoroughly cleaned, the purity of the material must be confirmed by at least two independent methods. The most commonly used polyacrylamide gel electrophoresis (PAGE) and ultracentrifugation. After protein purification, it is divided into two to three or more parts. Each part is treated with different protease enzymes (trypsin, chymotrypsin) or reagents (bromine cyan, iodobenzoic acid). The result is two to three (or more) sets of polypeptides (protein segments). Special purity requirements are imposed on proteinase enzymes, otherwise the subsequent determination of the sequence of alternation of segments of peptides in the native protein chain will be difficult. Of particular difficulty is the recognition of places of disulfide bridges between cysteine \u200b\u200bresidues. The resulting mixture of peptides is separated by electrophoresis, after which it is possible to start the sequencing procedure directly. The length of a single peptide should not exceed 40 AK residues.
The most commonly used method for sequencing peptides (establishing the sequence of AK residues in them) is the P. Edman procedure, using phenylisothiocyanate (FITZ). The procedure underlies the operation of automatic sequencers. A sample of the purified peptide is applied to the surface of the reaction vessel in the form of a film. Often, the peptide is covalently sutured from the end of the free COOH group with the surface material of the reaction vessel. Then repeat the cycles from a series of reactions. One series of reactions includes:
- the interaction of the FITC with the terminal AK residue having a free NH 2 group, the so-called FTK- ( phenylthiocarbamoyl)-derivative:
- excess FITC is removed, the pH of the medium is changed by adding heptafluorobutyric acid, with the conversion of FTK to FTG ( phenylthiohydantoin)-derivative:
- The FTG derivative of the amino acid is removed from the reaction medium by extraction with 1-chlorobutane and the series of reactions is repeated in the next cycle.
In one cycle, one amino acid residue is removed from the NH2 edge of the peptide. Since reactions with FITC do not proceed quantitatively, but at best, by 95 percent, interfering factors gradually accumulate - FTG derivatives from AK residues that did not react to previous cycles. In the most favorable cases, it is possible to reliably identify the sequence of only about 40 AK residues. However, due to the automation of the process, the work is still substantially facilitated.
INTERPRETATION WITH PEPTIDASES:
a) from the N-terminal residues using AMINOPEPTIDASES ( chromatographic identification and kinetics of accumulation of the corresponding AKs.
b) from the C-ends with carboxypeptidases (similarly).
HYDRAZINOLYSIS (in an anhydrous medium at 100 degrees C), with the exception of the last residue with free COOH, all turn into acid hydrazides:
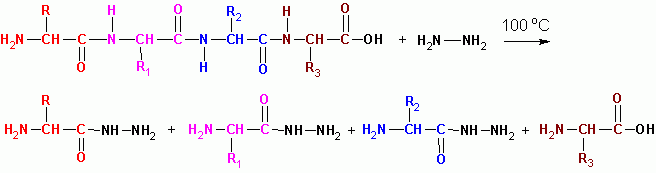
The sequence of peptides in protein molecules is determined by overlapping fragments of peptides:
G-w-v-r A-o-v-k C-E-C-D tryptic peptides (trypsin)
G-w V-R-A-O V-K-C-E-C-D chymotriptic peptides (chymotrypsin)
RECOMMENDED LITERATURE:
1. Stroyev EA Biological chemistry. M., 1986.
2. Striyer L. Biochemistry, in 3 volumes M., Mir, 1984.
3. White, Handler, Smith and others. Fundamentals of biochemistry, in 3 vols. M., Mir, 1981.
4. Ovchinnikov Yu.A. Bioorganic chemistry, M., Enlightenment, 1987.
5. Anisimov et al. Fundamentals of biochemistry. M., High School, 1986.
One of the features of proteins is their complex structural organization. All proteins have a primary, secondary and tertiary structure, and those that have two or more PPCs in their composition also have a quaternary structure (ES).
Primary Protein Structure (PSB) – this is the alternation order (sequence) of amino acid residues in the PPC.
Even proteins of the same length and amino acid composition can be different substances. For example, two different dipeptides can be made up of two amino acids:
With the number of amino acids equal to 20, the number of possible combinations is 210 18. And if you take into account that in the PPC each amino acid can occur more than 1 time, then the number of possible options is difficult to calculate.
Determination of the primary structure of the protein (PSB).
PSB proteins can be determined using phenylthiohydantoin
method
. This method is based on the reaction of interaction. phenylisothiocyanate
(FITZ) with α-AK. The result is a complex of these two compounds - FITZ-AK
.
For example, consider a peptide ![]() in order to determine its PSB, that is, the sequence of the connection of amino acid residues.
in order to determine its PSB, that is, the sequence of the connection of amino acid residues.
FITC interacts with terminal amino acid (a). The complex is formed FTG-a, it is separated from the mixture and the authenticity of the amino acid is determined but. For example, this is - asn
![]() etc. All other amino acids are sequentially separated and identified. This is a time consuming process. The determination of medium-sized PSB protein takes several months.
etc. All other amino acids are sequentially separated and identified. This is a time consuming process. The determination of medium-sized PSB protein takes several months.
The priority in decoding PSB belongs Senger (1953), which opened the insulin PSB (Nobel Prize Laureate). The insulin molecule consists of 2 PPCs - A and B.
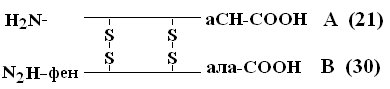
A-chain consists of 21 amino acids, chain B - of 30. Between themselves, PPC are connected by disulfide bridges. The number of proteins whose PSB has been determined by now reaches 1,500. Even small changes in the primary structure can significantly change the properties of the protein. Red blood cells of healthy people contain HbA - when replaced in the цепи-chain of HbA, in the 6th position gloom on shaft a serious illness occurs sickle cell anemiain which children born with this anomaly die at an early age. On the other hand, variations in PSB are possible that do not affect its physicochemical and biological properties. for example, HbC contains in the 6th position of the b-chain instead of glucose, HbC almost does not differ in its properties from HbA, and people who have such Hb in red blood cells are practically healthy.
PSB stability provided mainly by strong covalent peptide bonds and, secondly, by disulfide bonds.
The secondary structure of the protein (VSB).
PPV proteins have great flexibility and acquire a certain spatial structure or conformation. In proteins, 2 levels of such a conformation are distinguished - these are VSB and tertiary structure (TSB).
Fab – this is the configuration of the PPC, that is, the way it is laid or twisted into some kind of conformation, in accordance with the program laid down in PSat
Three main types of VSD are known.:
1) -spiral;
2) b-structure (folded layer or folded sheet);
3) messy tangle.
-spiral .
Her model was proposed by W. Pauling. It is most likely for globular proteins. For any system, the most stable is the state corresponding to the minimum free energy. For peptides, this state occurs when the CO– and NH– groups are joined together by a weak hydrogen bond. AT a spirals NH– groups of the 1st amino acid residue interact with the CO – group of the 4th amino acid. As a result, the peptide backbone forms a spiral, for each turn of which 3.6 AA residues fall.
1 helix step (1 turn) \u003d 3.6 AK \u003d 0.54 nm, rise angle - 26 °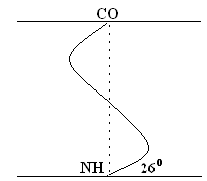
The twisting of the PPC occurs clockwise, that is, the spiral has the right move. Every 5 turns (18 AK; 2.7 nm), the PPC configuration is repeated.
Is stabilized Fab primarily hydrogen bonds, and secondly, peptide and disulfide bonds. Hydrogen bonds are 10-100 times weaker than ordinary chemical bonds; however, due to their large number, they provide a certain rigidity and compactness of the VSB. The lateral R-chains of the a-helix are facing outward and are located on different sides from its axis.
b -structure .
These are the folded sections of the PPC, resembling a leaf folded in an accordion in shape. The PPC layers can be parallel if both chains begin at the N– or C – end.
If adjacent chains in a layer are oriented by opposite ends N – С and С – N, then they are called antiparallel.
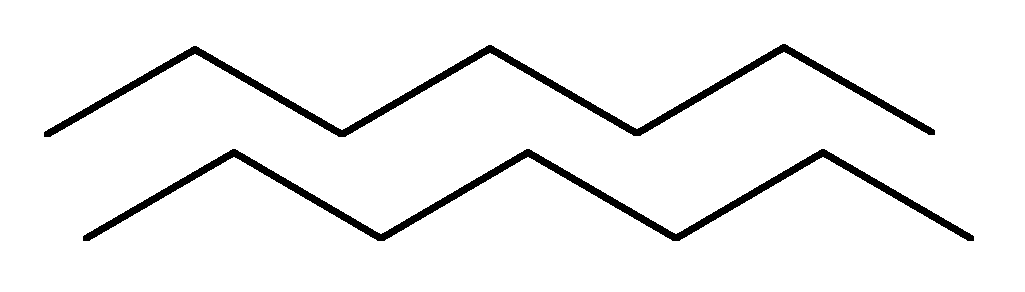 parallel
parallel
antiparallel
The formation of hydrogen bonds occurs, as in the a-helix, between CO– and NH– groups.
Determination of the primary structure of proteins
The determination of the primary structure is preceded by denaturation and breaking of transverse disulfide bonds in the protein. This is achieved through an excess of mercaptoethanol.
Cystine is converted into two cysteine \u200b\u200bresidues, which are then blocked with an excess of iodoacetic acid to prevent the reverse formation of S-S- bonds.
The cleavage of the polypeptide chain into fragments is usually carried out using proteolytic enzymes, such as trypsin, chymotrypsin or pepsin. These enzymes act on different parts of the polypeptide chain, as they have an increased affinity for various amino acid residues. Necessary amino acid residues, i.e. spatial environment of the attacked peptide bond. It turned out that trypsin hydrolyzes only those peptide bonds in the formation of which the carboxyl group of lysine or arginine is involved, and chymotrypsin hydrolyzes bonds according to phenylalanine, tryptophan and tyrosine. Typically, proteolytic enzymes that hydrolyze polypeptide chains are pre-immobilized on insoluble matrices to more easily separate them from hydrolysis products. The amino acid sequences of each polypeptide fragment are then determined. For this, the Edman method is most often used, consisting in the analysis of a polypeptide only from the N-terminus. The terminal amino acid, when reacted with phenylisothiocyanate in an alkaline medium, forms a stable compound that can be cleaved from the polypeptide without degradation. The phenylthiohydantoin (FTG) derivative of an amino acid is identified by a chromatographic method.
After identification of the terminal N-amino acid residue, the label is introduced into the next amino acid residue, which becomes the terminal. Edman's method can be automated using a sequencer (from the English. sequetice -sequence) by which the FTG derivatives are cleaved from the polypeptide and identified by high performance liquid chromatography.
F. Sanger for the first time completely deciphered the primary structure of the protein hormone insulin using the Edman method.
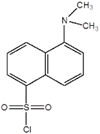
Another highly sensitive method is the so-called dansyl method associated with the addition of dansyl chloride (1-dimethylamino-naphthalene-5-sulfonyl chloride) to the terminal amino acid according to the following scheme:
The primary structure of the protein can be established indirectly as follows: first, the corresponding cDNA is obtained., Then the clone related to the analyzed protein is identified, and the primary structure of the protein is determined by the alternation of nucleotides in it using a library of amino acid sequences.
Determination of the secondary structure of proteins
Optical methods are mainly used to determine the secondary structure of proteins. Of course, the X-ray diffraction method is more reliable, but its application is fraught with certain difficulties and requires considerable time. Optical methods such as dispersion of optical rotation and circular dichroism are simpler and, very importantly, can determine changes in the secondary structure of the protein in solutions. Using the dispersion of optical rotation, one can obtain information on the degree of helicity of a protein macromolecule. Despite the fact that the method is approximate, spiral-coil transitions are quite clearly visible. As for the method of circular dichroism, its spectrum is determined by the set of angles w and q, characteristic of one or another type of secondary structure. Both methods can be regarded as screening, and for complete identification of the secondary structure, they must be combined with x-ray analysis of proteins.
Proteins are high molecular weight organic compounds, monomeric units, which are α-amino acids, interconnected by a peptide bond. Classification:
- in the form of molecules (globular or fibrillar); Fibrillar proteins. Polypeptide chains are parallel to each other, elongated chains form long strands or layers. They serve as the main organic material of connective tissue and are a structural component of muscle fiber. For example, collagen, elastin. Globular proteins. Polypeptide chains are tightly folded into compact spherical structures (globules). They are water soluble. For example, enzymes.
- by molecular weight (low molecular weight, high molecular weight, etc.);
- chemical structure (presence or absence of non-protein part);
- on the functions performed (transport, protective, structural proteins, etc.);
- by localization in the cell (nuclear, cytoplasmic, lysosomal, etc.);
- by localization in the body (blood proteins, liver, heart, etc.);
Primary Protein Structure Is a sequence of amino acid residues in a polypeptide chain of a molecule connected by peptide bonds.
The peptide bond is characterized by the following properties:
1) the O and H atoms of the peptide bond are transoriented;
2) four atoms of the peptide bond lie in the same plane, i.e. peptide bonds are characterized by coplanarity;
3) the peptide bond in the protein molecule exhibits keto-enol tautomerization;
4) the length of the C-N bond, equal to 0.13 nm, has an intermediate value between the length of the double covalent bond (0.12 nm) and the single covalent bond (0.15 nm), which implies that rotation around the C-N axis is difficult;
5) the peptide bond is stronger than the usual covalent bond, because it is one and a half (due to the redistribution of electron density).
For each individual protein, the amino acid sequence in the polypeptide chain is unique. It is genetically determined and in turn determines higher levels of organization of a given protein.
The amino acid residue located at the end of the chain where there is a free amino group is called the amino terminal, or N-terminal, and the residue at the other end, bearing the free carboxyl group, is called a carboxy-terminal or C-terminal. The name of the polypeptide begins at the N-terminus.
Fragmentation of polypeptide chains
Chemotrypsin cleaves peptide bonds formed by carboxy groups of aromatic AA
T trypsincleaves peptide bonds formed by AA containing two amino groups (lysine, arginine)
Bromocyan (CNBr) cleaves peptide bonds formed between the carboxyl group of methionine and the amino group of any other AK.
Hydroxylamine (NH2OH), cleaves peptide bonds formed between residues of aspartic acid and glycine
N-bromosuccinamide cleaves peptide bonds formed between the carboxyl group of tryptophan and the amino group of any other AK).
Sequinators - automatic devices that allow using the Edman method (reaction of polypeptides with phenylisothiocyanate) to study the primary structure of peptides containing up to several tens of amino acid residues.
Phenylthiohydantoin method A method for determining the primary structure of a protein. Based on the interaction of phenylisothiocinate with the N-terminal AK. The essence of the method is to process the studied peptide with a specific set of reagents, which leads to the elimination of one amino acid from the N-end of the sequence. Cyclic repetition of the reaction and analysis of reaction products provide information on the sequence of amino acids in the peptide.
Methods for studying the structure of proteins
Complete hydrolysis in either alkaline or acidic conditions.
Acid hydrolysis is carried out for 24 hours at t \u003d 110 6-NHCl. The hydrolysis products are separated by ion exchange chromatography (on a column) with sulfonated polystyrene. Amino acid fractionation: using the ninhydron, the amount of amino acid is determined by color. Then, each amino acid is washed and photometrically determined. In this way, light amino acids (fingerprints) can be identified. If there is a small amount of protein 10 -9, then fluorescamine reagent is used.
Determination of amino acid sequence.
Make sure that the chain is one.
1.Senger method:defin. one AK with N-terminus
Can use dansyl chloride
(as well as in the previous district only for chlorine)
2. Edman's method - allows you to determine dozens of amino acids from the N-terminus
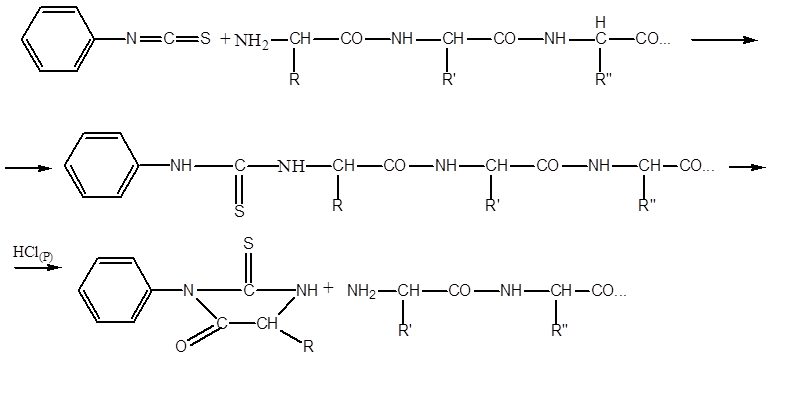
Edman designed the sequencer. the protein is placed as a thin film in a rotating cylinder. the vessel where it undergoes cleavage Deciphered 60 amino acids from the N-terminus of the whale myoglobin.
3. Definition of the 1st amino acid from the end of hydrazine.
4. The carboxypeptidase enzyme can be sequentially cleaved at the 1st amino acid from the C-terminus.Used to determine small polypeptides (up to 100)
If more than 100.
Trepsin- cleaves peptide bonds formed with the C-terminus of basic amino acids - lysine and argenin.
Chemotrepsin- cleaves the peptide bond from the end of aromatic and dicarboxylic acids.
Separated chromatographically, begin to determine. Again take a portion of the polypeptide. Cleaved in another way, using a chemical reagent, use cyanogen bromide - a cleavage from the C-terminus of methylamine. Look, overlapping sections are compared.
Hydroxylamine–NH 2 OH- cleaves the asparagine-glycine bond.
2-nitro-5thiocyanobenzoate - cleaves where the amino group of cystoine.
1953 - insulin is the very first sequence. Immunoglobulin (4 polypeptide chains) is the largest 1300 amino acid residues.
In proteins, all amino acids can be determined.
Fingerprint method
Bromcian cleaves methiamine from the C-terminus (90%HCOOH)
Sequencing is needed for:
1) To identify the molecular basis of biol. activity.
2) How the primary structure affects the second, tertiary and quaternary.
3) A change in sequence can lead to a disease (hereditary).
4) Protein sequence data can tell a lot about evolution.
2 RNA
RNA- H 3 PO 4; Ribose Adenine; Guanine; Cytosine; Uracil
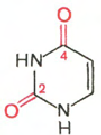
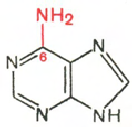
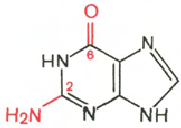
Cytosine Uracil Adenine Guanine
Carbohydrates (ribose and deoxyribose) in DNA and RNA molecules are in β-D-ribofuranose form:
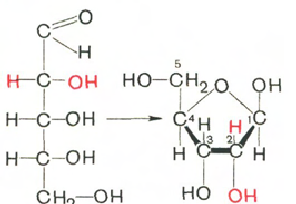
D-ribose β-D-ribofuranose
There is no exact data on the amount of RNA, since its content in different cells is largely determined by the intensity of protein synthesis. RNA accounts for about 5–10% of the total cell mass. The current classification of various types of cellular RNA is based on data from topography, function, and molecular weight. Three main types of RNA are distinguished: matrix (informational) - mRNA, which makes up 2-3% of all cellular RNA; ribosomal - rRNA, comprising 80–85% and transport - tRNA, which is about 16%. These 3 species differ in nucleotide composition and functions.
Matrix RNA formed during transcription. It carries an exact copy of the genetic information encoded in a specific section of DNA, namely about the sequence of amino acids in proteins.
Matrix RNA (mRNA) is synthesized in the nucleus on a DNA matrix, then enters the ribosome, performing a matrix function in protein synthesis. When entering from the nucleus into the cytoplasm, mRNA forms complexes with specific RNA-binding proteins - the so-called informosomes capable of reversible dissociation. Informosomes are considered as a transport form of mRNA that promotes the formation of polyribosomes in the cytoplasm.
Transport RNA
(tRNAs) have a small molecular weight and are contained in the soluble fraction of the cytoplasm, performing the function of transferring amino acids to the site of protein synthesis - the ribosome. The tRNA molecule is a single polynucleotide chain twisted "on itself." All tRNAs are built according to one plan, all of them fit into the clover leaf model. The main principle is the formation of the maximum number of hydrogen bonds between nitrogenous bases. The clover leaf contains five spiral stems, four of which end in loops of unpaired nucleitides. In the center of the molecule is an uncoiled region. 3 ’and 5’ - the ends of the polynucleotide chain are paired to form an accepting stem. Resists an accepting stem anti-codon stem. It carries an anti-codon loop, which consists of 7 nucleotides. The anticodon loop contains in its middle part an anticodon consisting of 3 nucleotides complementary to the codon of a given amino acid in mRNA. The pseudouridine loop carried by the T-stem contains the minor component pseudouridine. It consists of 7 nucleotide residues. It is believed that it is with this loop that tRNA interacts with the ribosome. D– the stem carries a loop of 8-12 nucleotides. This is a loop of dihydrouridine, it always contains several residues of the minor component of dihydrouridine. 
Ribosomal RNA - the basis on which the proteins are located, forming a ribosome. Ribosomes are localized mainly in the cytoplasm, in addition, in the nucleus of mitochondria and chloroplasts. The nucleotide composition is similar: guanyl nucleotides predominate, uridyl and cytidyl ones are in small quantities, there are no minor bases. In size and molecular weight, ribosomes are divided:
Bacterial ribosomes (relatively small). Sedimentation constant 70S.
Ribosomes of eukaryotic cells. Sedimentation constant 80S.
Ribosomes of mitochondria and chloroplasts.
In the hydrolysis of RNA ribose, adenine, guanine, uracil, cytosine, phosphoric acid are formed.